
Abstract
Abstract
A large number of studies demonstrated the importance of different HIV RNA structural elements at all stages of the viral life cycle. Nevertheless, the significance of many of these structures is unknown, and plausibly new regions containing RNA structure-mediated regulatory signals remain to be identified. An important characteristic of genomic regions carrying functionally significant secondary structures is their mutational robustness, that is, the extent to which a sequence remains constant in spite of despite mutations in terms of its underlying secondary structure. Structural robustness to mutations is expected to be important in the case of functional RNA structures in viruses with high mutation rate; it may prevent fitness loss due to disruption of possibly functional conformations, pointing to the specific significance of the corresponding genomic region. In the current work, we perform a genome-wide computational analysis to detect signals of a direct evolutionary selection for strong folding and RNA structure-based mutational robustness within HIV coding sequences. We provide evidence that specific regions of HIV structural genes undergo an evolutionary selection for strong folding; in addition, we demonstrate that HIV Rev responsive element seems to undergo a direct evolutionary selection for increased secondary structure robustness to point mutations. We believe that our analysis may enable a better understanding of viral evolutionary dynamics at the RNA structural level and may benefit to practical efforts of engineering antiviral vaccines and novel therapeutic approaches.
1. Background
H
In addition to being a virus of an immense clinical significance, HIV possesses important biophysical properties making it especially appealing for researchers. An extremely high mutation rate (Cuevas et al., 2015), a complex coding capacity despite its relatively small genome size (Frankel and Young, 1998; Hryckiewicz et al., 2011), and a variety of expression regulation mechanisms and codes such as frameshifts, alternative splicing, and secondary structures (Jacks et al., 1988; Malim et al., 1989; Cullen, 1991; Parkin et al., 1992; Damgaard et al., 2004; Seelamgari et al., 2004; Goff, 2007; Stoltzfus, 2009; Brakier-Gingras et al., 2012; Karn and Stoltzfus, 2012; Schiralli Lester and Henderson, 2012), together with availability of reachable experimental data (e.g., Shankarappa et al., 1999; Liu et al., 2006; Watts et al., 2009; Zanini et al., 2015) and high-quality annotated genomic sequences (“HIV Databases,” n.d.), turn HIV into a good model system for studying viral evolution and replication (Shankarappa et al., 1999; Liu et al., 2006; Zanini and Neher, 2013).
HIV and other viruses undergo a rapid evolutionary selection that enables them to evade the host immune system and to efficiently compete with endogenous transcripts of the host cell over the gene expression machinery. Mechanisms that facilitate an efficient and selective viral replication are inherent in the nucleotide composition of the viral genomic sequence itself and can involve the recruitment and/or modification of specific host factors. Nonsynonymous mutations that alter the amino acid sequence provide a distinct evolutionary advantage due to selective pressure, allowing viruses to escape from innate defense mechanisms and acquired immune surveillance of the host, and to rapidly adapt to new cell types, tissues, or species. Yet, genomes (and even coding sequences), both viral and of other organisms, not only code for protein products but also carry additional information encrypted in the composition of alternating codons (Lavner and Kotlar, 2005; Parmley and Huynen, 2009; Tuller et al., 2010; Plotkin and Kudla, 2011; Li et al., 2012; Zur and Tuller, 2012; Mortimer et al., 2014; Tuller and Zur, 2014; Goz and Tuller, 2015). Being related to different biophysical and evolutionary characteristics, this information can be induced by synonymous mutations, which preserve the underlying protein, and may play an important regulatory role in different viral replication stages.
In particular, an important feature of viral RNA, which is encoded in synonymous (and nonsynonymous) aspects of its nucleotide composition, is its structure/conformation that is formed by the molecule folding upon itself as a result of hydrogen bonds and other biochemical connections acting between pairs of nucleotides. The number and strength of these bonds determine the minimum free folding energy (MFE), which is related to the folding strength of a structure; more negative MFE is related to possibly stronger and more stable folding, whereas less negative MFE corresponds to weaker and less structured conformations.
A large number of experimental and computational studies in HIV and other viruses and organisms demonstrated the importance of different RNA structural elements at all stages of the viral life cycle. Among others, these elements were found to be involved in transcription activation, reverse transcription initiation, facilitation of genomic dimerization, direction of HIV packaging, manipulation of reading frames, regulation of RNA nuclear export, regulation of translation and replication, polyadenylation signaling, and interactions with viral and host proteins (Malim et al., 1989; Cullen, 1991; Abbink and Berkhout, 2003; Damgaard et al., 2004; D'Souza and Summers, 2005; Gaudin et al., 2005; Moore and Hu, 2009; Stoltzfus, 2009; Watts et al., 2009; Karn and Stoltzfus, 2012). An RNA secondary structure model for the complete HIV genome has recently been published based on SHAPE technology (Watts et al., 2009); as a result, several well-known RNA structures were confirmed, and numerous new structured motifs were proposed. Nevertheless, the significance of many of these structures is unknown, and plausibly, new regions containing RNA structure-mediated regulatory signals remain to be identified.
An important characteristic of genomic regions carrying functionally significant conformations is their mutational robustness, that is, the extent to which the phenotype associated with the secondary structure remains invariant despite mutations. In the light of the extremely high error rate in the process of HIV replication, robustness may protect viruses in the face of deleterious mutation and prevent fitness loss due to disruption of possibly functional secondary structures.
Several previous studies have addressed the evolution of secondary structure robustness in viruses and other organisms. For example, Borenstein and Ruppin (2006) provided an evidence of a direct evolutionary selection of mutational robustness in micro RNA molecules, whereas Tuller et al. (2011) suggested a signal of mutational robustness at the beginning of the mRNA. In Wagner and Stadler (1999), it was shown that conserved RNA secondary structure elements are consistently more robust than nonconserved elements in different groups of RNA viruses (in particular, trans-activation response element (TAR) and Rev responsive element (RRE) elements of HIV were analyzed). In Churkin et al. (2010), the authors demonstrated the direct evolution of mutational robustness in the functionally important structural elements of the Hepatitis C virus (HCV); in Waldispühl et al. (2008), the structure robustness of HCV cis-acting replication element and HIV TAR was investigated.
In the current work, we performed for the first time, a genome-wide computational analysis of HIV coding regions aiming to identify significant signals encoded by synonymous information that are related to evolutionary selection for strong mRNA folding. Further, we focused on sites spanned by the predicted, evolutionarily significant, and local structural elements to analyze whether in addition to being selected for stability, their structures also undergo a direct selection pressure toward increased mutational robustness.
The study in Wagner and Stadler (1999), included, among others, the analysis of two HIV elements (TAR and RRE) and was based on statistical comparison of conserved and not conserved regions from the viral genome; in contrast, we analyzed the selection for a structural robustness on predicted and significantly stable sites within the coding regions, basing on randomization models inspired by the previous studies on other biological entities (Borenstein and Ruppin, 2006; Churkin et al., 2010). These randomization models exclude the possibility of an implicit enhancement of robustness as a side effect of specific sequence (e.g., amino acid content and order of the viral proteins) and structure (e.g., the strength of the RNA folding) characteristics, and permit to reveal statistically significant signals of a direct selection pressure. HIV robustness was also addressed in Waldispühl et al. (2008) by computing energetically favorable multiple-point mutations and studying their impact on the robustness of the secondary structure; however, this study analyzed only the TAR noncoding element and did not make use of a null model to provide the statistical significance of the reported findings and to control for factors that could have a roundabout effect on robustness (as mentioned above).
We believe that our analysis will enable a better understanding of viral evolutionary dynamics at the structural level and may benefit practical efforts of engineering antiviral vaccines and novel therapeutic approaches.
2. Results
The different general stages of the study appear in Figure 1 (see Section 4 for additional details). HIV coding regions of the HXB2 HIV strain were prepared according to published annotations (Fig. 1A I). To analyze the evolutionary selection for strong folding in local genomic elements, we used a reference model which promises that signals of increased structural stability cannot be explained by some of the fundamental properties of the wild-type sequences— the amino acid composition of the encoded proteins and/or disrupted stacking base-pairs (dinucleotide and amino acid preserving model). To this aim, we designed 1000 randomized variants that preserved both the encoded protein and the distribution of frequencies of pairs of adjacent nucleotides (dinucleotides) known to have a significant impact on the folding strength (Zuker and Stiegler, 1981; Nussinov, 1984; Workman, 1999) (Fig. 1A II).
MFE was estimated in all genomic windows of length 150 nt within each wild-type coding region and its randomized variants, and the resulting values were used to construct local MFE profiles; each position in a profile contained an MFE value computed in a window starting at this position (Fig. 1A III, B).
To identify positions along the coding regions that were possibly selected during the course of viral evolution for significantly strong folding (i.e., more negative MFE), we investigated the positionwise statistical differences between the MFE profiles corresponding to the wild-type sequences and MFE profiles of their randomized variants (Fig. 1A IV). For each coding sequence, we identified positions for which the MFE values were found to be lower than in more than 99% of the corresponding randomized variants (i.e., positions with empiric MFE-associated p-value <0.01) with the Benjamini–Hochberg false discovery rate (BH-FDR) of 1%; regions spanned by these positions have significantly less negative MFE in comparison to random and are conjectured to undergo an evolutionary selection for strong folding (for convenience we call them MFE-selected positions/regions).
To investigate which of the MFE-selected regions have also evolved an increased robustness to mutations associated with its secondary structure, we first defined the structure-based mutational robustness (SMR) measure as an extent to which single-point mutants tend to modify the secondary structure of a region (Fig. 1C, D). The SMR takes values between 0 and 1; the higher the value, the higher is the robustness.
To infer a direct selection for SMR of specific genomic regions and exclude the effect of intrinsic robustness associated with the increased stability of the secondary structure (strongly affected by stacking pairs of nucleotides) and/or the selection for amino acid order and/or content, we used the aforementioned dinucleotide and amino acid preserving randomization model (Fig. 1V).
In addition, to provide the evidence that the increased structural robustness to mutations has evolved independently and not as a side effect of the evolution of specific conformations (certain conformations may be inherently more robust than others), it was essential to demonstrate that a native secondary structure is significantly more robust than phenotypically similar randomized variants. To this aim, we used a second randomization model that controls for the effect of the specific conformation by generating random sequences with exactly the same structure as in the wild type (but not necessary preserving the encoded peptide) (Fig. 1VI).
These randomization models aimed at providing an initial answer to the question whether the SMR values of the analyzed sites can be explained by nondirect factors, other than mutational robustness, such as the selection for specific secondary structure, the selection for folding strength, and/or amino-acid compositions.
For every MFE-selected region, we computed the SMR measure for wild-type coding sequences and their variants, generated by each one of the randomization models (1000 variants per model; Fig. 1VII). The wild-type and randomized SMR values were then compared (for each region and randomization model separately); a region, for which the wild-type SMR was found to be higher than in 95% of the variants (SMR-associated p-value <0.05) for both randomization models (Fig. 1VIII), was hypothesized to undergo a direct evolutional selection for SMR.
In the following section, we present a detailed analysis of our findings. More details about the methods used in the study can be found in Section 4.
2.1. Evidence that specific regions of HIV structural genes undergo an evolutionary selection for strong folding
In each one of the HIV structural genes (gag, pol, and env), one cluster of MFE- selected positions was identified. In env and pol genes, these clusters overlap with the RRE and the ribosomal frameshift site, both known to contain important functional secondary structures (Gaudin et al., 2005; Fernandes et al., 2012).
To the best of our knowledge, the cluster identified in gag gene does not correspond to any known functional site and may play important (yet unknown) roles in the viral life cycle.
Table 1 summarizes our findings.
The start–end positions of identified clusters of MFE-selected positions, and start–end positions of known functional sites (as given in the references) overlapping with these clusters are specified. The cluster start position records the start position of the first 150 nt MFE-selected window in the cluster; the cluster end position records the end position of the last 150 nt MFE-selected window in the cluster. All coordinates are given with respect to the HXB2 reference genome. We have not found in the literature any known functional site that corresponds to the predicted cluster in gag (the site coordinates are unspecified and therefore marked by “?”
MFE, minimum free folding energy; RRE, Rev responsive element.
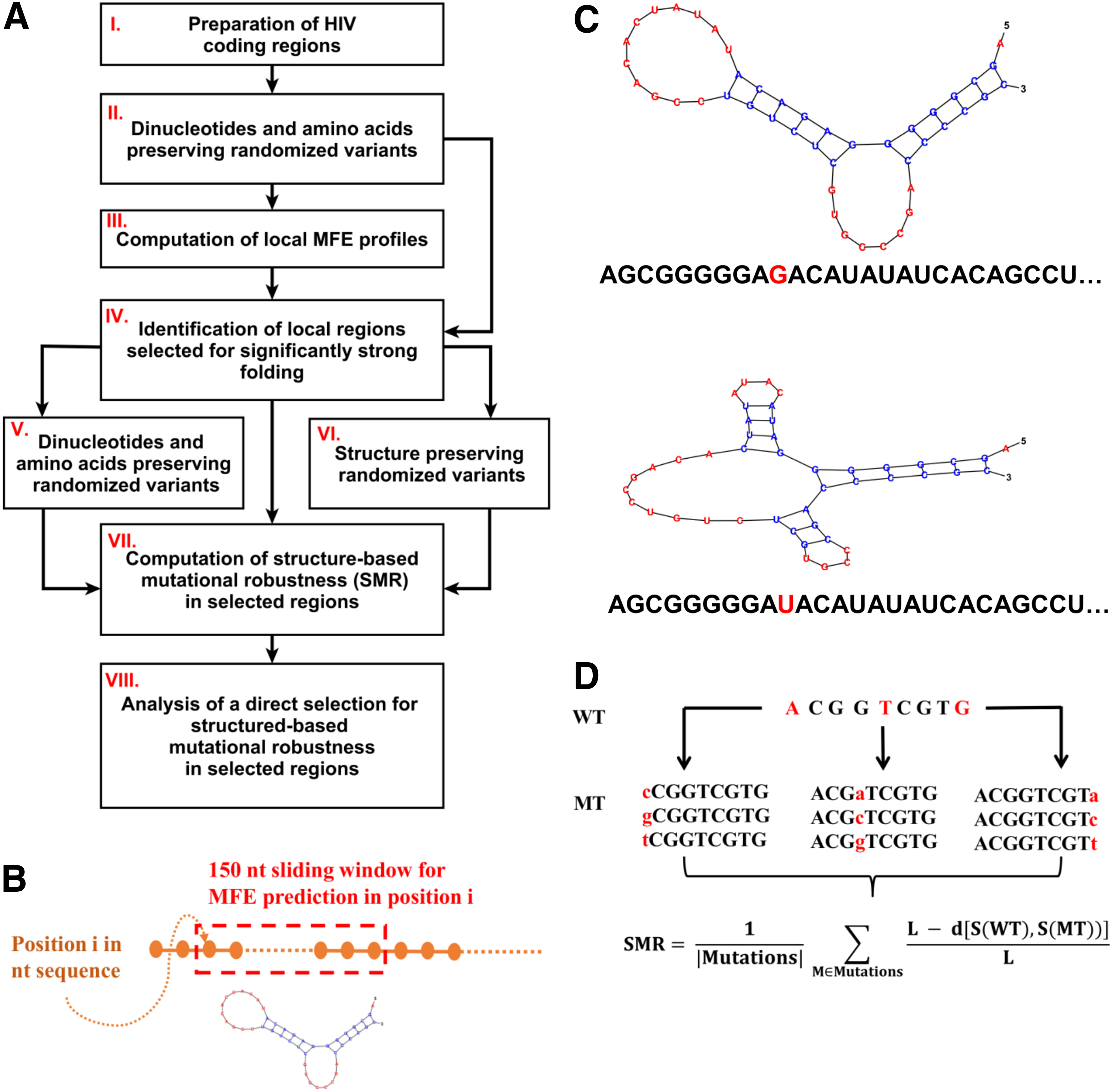
In the remaining seven nonstructural genes, we found no statistical evidence of evolutionary selection for strong folding within their coding regions; although several positions where the MFE was more negative than in 99% of the randomized variants (p-value < 0.01) were detected, none of them passed the BH-FDR correction (Fig. 2).
Evidence that specific regions of HIV structural genes undergo an evolutionary selection for strong folding. Each panel corresponds to wild-type (blue) and mean randomized (green) MFE profiles for one gene. The y-axis corresponds to the MFE (kcal/mol) in the 150 nt genomic window starting at positions specified along the x-axis (nucleotide coordinated given with respect to the start of the coding region); red points—positions with MFE-related p-value <0.01 (in these positions, the wild-type folding is stronger than in 99% of randomized variants); yellow points—MFE-selected positions (MFE p-value <0.01 & BH-FDR = 0.01), these positions span genomic regions that are conjectured to undergo an evolutionary selection for strong folding. We can see clusters of MFE-selected positions in each one of the structural genes (env, gag, pol); in other genes, no evidence of selection for strong folding was found.
2.2. HIV Rev responsive element undergoes a direct evolutionary selection for increased secondary structure robustness to point mutations
Next, we were interested to check whether the significantly structured regions corresponding to the identified clusters of MFE-selected positions (Table 1) also undergo a direct evolutionary selection for robustness of their secondary structures with mutations in the underlying sequence.
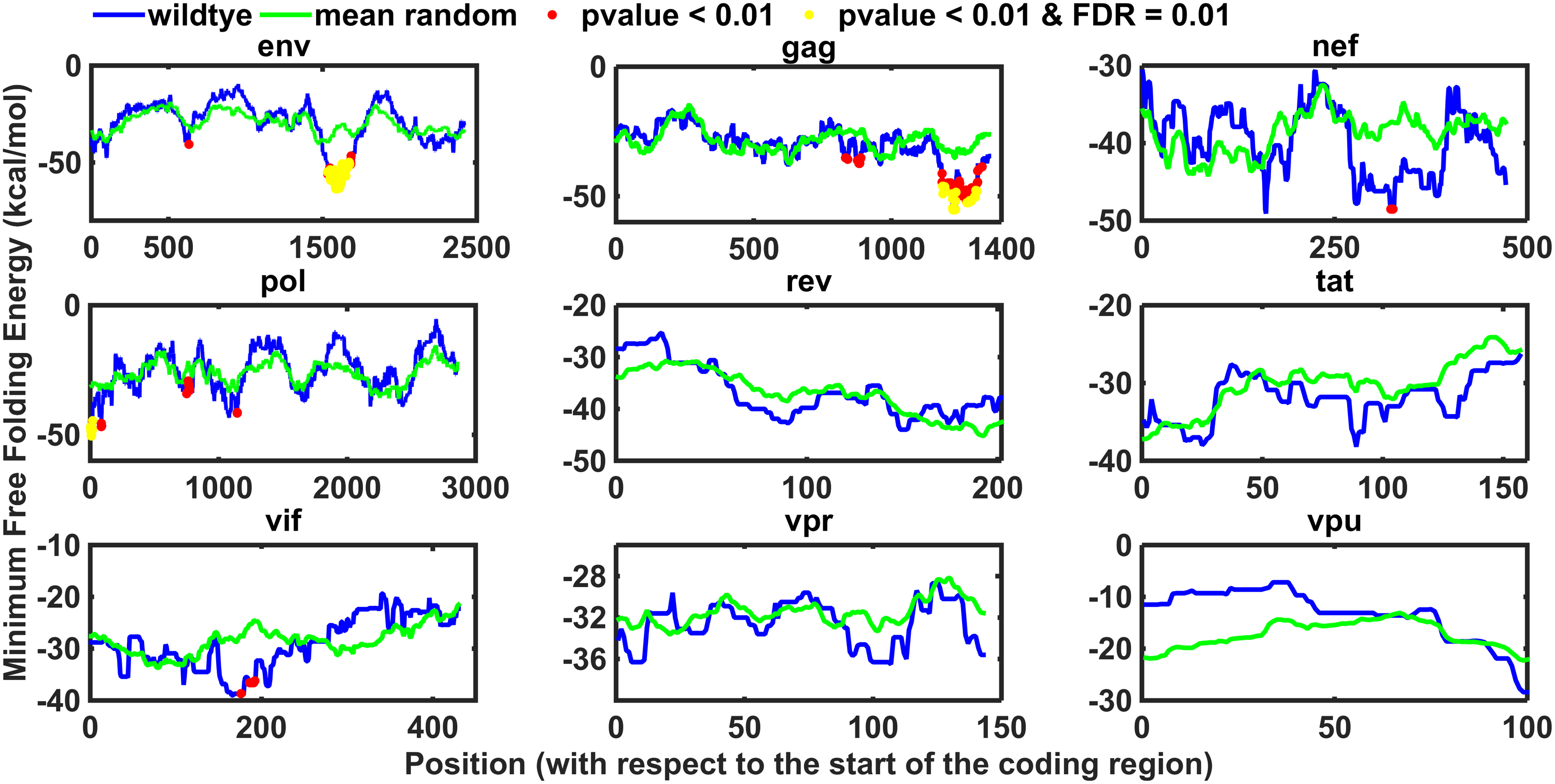
In Figure 3, we can see that RRE was found to be significantly more robust than in random (p-value = 0.028, z-score = 1.6 with respect to the structure preserving randomized variants; p-value = 0.013, z-score = 1.2 with respect to the dinucleotide and amino acid preserving models), and this robustness cannot be explained by the specific secondary structure of the corresponding region, its folding strength, and/or other sequence attributes, such as composition of dinucleotides and amino acids. Thus, we can conclude that our analysis supports the conjecture that the RRE secondary structure undergoes a direct evolutionary selection for mutational robustness.
Structure-based mutation robustness of RRE. X-axis—variant id: 1, wild type; 2, 1001–randomized. Y axis—SMR (see Section 4). The red line corresponds to the wild-type SMR value. The p-value (portion of randomized variants with a higher robustness than in wild type) and z-score (number of standard deviation of the wild-type SMR that is higher than the mean randomized SMR) are specified in red. Results for structure preserving
In contrast, we have not found a significant robustness signal for the other two selected sites in pol and gag genes (data not shown).
3. Discussion
We have shown for the first time that previously known functional genomic elements, RRE and gag-pol ribosomal frameshift site, seem to undergo an evolutionary selection for strong local mRNA folding.
RRE is a conserved and highly structured element that consists of ∼350 nucleotides spanning the border of gp120 and gp41 proteins in env coding region. It is a necessary regulatory factor for HIV expression and serves as a binding region on which molecules of the viral protein Rev assemble. The Rev-RRE complex promotes the export of viral mRNAs containing RRE from the nucleus to the cytoplasm, where they are both translated to produce new proteins and serve as a genomic material for new viral particles.
In agreement with the studies that showed that both RRE sequence and structure are necessary for proper functionality (Zapp and Green, 1989; Olsen et al., 1990; Fernandes et al., 2012), we have demonstrated that RRE not only undergoes an evolutionary selection for strong folding but its underlying secondary structure is also directly selected for an increased robustness to mutations.
Previous studies have demonstrated a correlation between the thermodynamic stability of RNA secondary structure and its mutational robustness (Wuchty et al., 1999); therefore, in general, it is plausible that the increased robustness may arise from the increased thermodynamic stability of the analyzed sequences. However, our randomization models, which control both the sequence characteristics affecting the folding strength and the underlying structure, support the conjecture that the discovered robustness has not evolved as a correlated side effect of sequence features, such as thermodynamic stability and/or encoded protein, but underwent a direct positive selection in the course of the viral evolution.
Another important site that has been predicted to undergo an evolutionary selection for strong folding was found to overlap with the beginning of the pol gene, taken to be the first T in the sequence TTTTTTAG. This sequence forms part of the secondary structure that potentiates ribosomal slippage on the RNA, resulting in the −1 frameshift and the translation of the Gag-Pol polyprotein (Jacks et al., 1988; Gaudin et al., 2005; Staple and Butcher, 2005; Low et al., 2014). Although we have shown that this secondary structure seems to undergo an evolutionary selection for strong folding, we found no evidence that it also undergoes an evolutionary pressure toward mutational robustness.
It can be speculated that the folding strength and not the RNA structure itself is important for controlling the efficiency of the frameshift. For example, in Mouzakis et al. (2013), the role of the secondary structure associated with frameshift was investigated and found to have a modest impact on frameshift efficiency, compatible with the hypothesis that the genomic secondary structure attenuates frameshifting by affecting the overall rate of translation. Consistently, in Ritchie et al. (2012), it was found that increased frameshifting efficiency was correlated with an increased tendency to form alternate and incompletely folded structures, suggesting a more complex picture of the role of the structure involving the conformational dynamics.
Finally, we have identified a previously unknown site in the gag gene with a highly significant signal of a strong mRNA folding, which can imply that this site may play an important functional role in the viral replication cycle. Although this site seems to be selected for a strong folding, our analysis revealed no evidence for a direct evolutionary selection for a structure-based robustness therein. Further studies are needed to explore the function of this element and its effect on the viral fitness. This can be done, for example, through a mutagenesis of this region followed with measurements of viral replication rates.
4. Methods
4.1. Data
The analysis presented herein was based on the HXB2 genome (GenBank accession number K03455). HXB2 was selected as the prototype, because it is the most commonly used reference strain for many different kinds of functional studies. All genomic coordinates/positions specified within this study were taken with respect to the start of the HXB2 genome. All the described computations were performed on each gene separately according to the annotated coordinates (“HIV Databases,” n.d.).
4.2. Local MFE profiles
The local MFE profiles were constructed by applying a 150 nt length sliding window to a genomic sequence; in each step, the MFE of a local subsequence enclosed by the corresponding window was calculated by Vienna (v. 2.1.9) package RNAfold function with default parameters (Lorenz et al., 2011). This function predicts the MFE and the associated secondary structure for the input RNA sequence, using a dynamic programming based on the thermodynamic nearest-neighbor approach (the Zucker algorithm) (Zuker and Stiegler, 1981).
4.3. Dinucleotides and amino acids preserving randomization
To model the composition of nucleotide pairs that are argued to have an important effect on formation of secondary structures, a model that preserves both the amino acid order and content and the frequency distribution of 16 possible pairs of adjacent nucleotides (dinucleotides) for each sequence separately was used. Although efficient methods exist for preserving the amino acid (e.g., permutation of synonymous codons) or the dinucleotide content [e.g., random generation of an Euler path in a De Bruijn-like graph, whose edges represent the dinucleotides (Altschul and Erickson, 1985)] separately, it has been difficult to combine them for satisfying both the constraints. To overcome these difficulties, we used an elegant algorithm proposed in Zhang et al. (2013), which is based on a multivariate Boltzmann sampling scheme, initially introduced in the context of enumerative combinatorics. This algorithm produces random variants that feature both correct dinucleotide frequencies and coding capacity, while being generated with provably uniform probability. We used the original source code, which can be found in http://csb.cs.mcgill.ca/sparcs
4.4. Structure-based mutational robustness
Following Borenstein and Ruppin, 2006, we define the SMR of an RNA sequence of length, L, as SMR = 〈(L−d)/L〉, where d is the base-pair distance between the secondary structure of the original sequence and the secondary structure of the mutant, averaged over all 3L one-mutant neighbors. The base-pair distance measures the dissimilarity between two structures as the number of closed pairs that are present in only one of the two structures (i.e., the number of pairs that should be opened or closed to transform one structure to the other). The SMR take values between 0 and 1; the higher the value, the higher is the robustness.
4.5. Structure preserving randomization model
To generate randomized sequences that fold into a given structure, we applied the inverse folding algorithm implemented in the RNAinverse function of Vienna package (Hofacker et al., 1994). Such inversely folded random reference set does not preserve the encoded protein, but perfectly retains the structural phenotype of the wild type.
Footnotes
Acknowledgment
We thank Dr. Richard Neher for very helpful discussions. E.G. is supported, in part, by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University. T.T. is partially supported by the Minerva ARCHES award.
Author Disclosure Statement
No competing financial interests exist.