
Abstract
Abstract
MicroRNAs (miRNAs) are a class of small noncoding RNAs that act as efficient post-transcriptional regulators of gene expression. In 2012, the first cross-kingdom miRNA-based interaction had been evidenced, demonstrating that exogenous miRNAs act in a manner of mammalian functional miRNAs. Starting from this evidence, we defined the concept of cross-kingdom functional homology between plant and mammalian miRNAs as a needful requirement for vegetal miRNA to explicit a regulation mechanism into the host mammalian cell, comparable to the endogenous one. Then, we proposed a new dedicated algorithm to compare plant and mammalian miRNAs, searching for functional sequence homologies between them, and we developed a web software called MirCompare. We also predicted human genes regulated by the selected plant miRNAs, and we determined the role of exogenous miRNAs in the perturbation of intracellular interaction networks. Finally, as already performed by Pirrò and coworkers, the ability of MirCompare to select plant miRNAs with functional homologies with mammalian ones has been experimentally confirmed by evaluating the ability of mol-miR168a to downregulate the protein expression of SIRT1, when its mimic is transfected into human hepatoma cell line G2 (HEPG2) cells. This tool is implemented into a user-friendly web interface, and the access is free to public through the website http://160.80.35.140/MirCompare
1. Introduction
M
The miRNA-mediated gene regulation may occur either through mRNA degradation or preventing mRNA from being translated. Plant miRNAs need high complementarity to recognize their substrate, and the target cleavage is considered the predominant pathway to repress gene expression. Several studies suggest that translational inhibition is also a common mechanism used by plant miRNAs to repress gene expression. In animals, the semicomplementary interaction leads to silencing achieved by translational repression (Mallory et al., 2004; Bartel, 2009; Huang et al., 2010; Liu et al., 2014; Xie et al., 2015). In 2013, Brennecke et al. (2005) described for the first time the minimal requirements for a functional miRNA:mRNA-target duplex in vivo. The study reported that any mismatch in positions 2–8 of the seed region strongly reduced the magnitude of the target regulation. Moreover, the minimal 5′ sequence complementarity necessary to confer target regulation is of four or five nucleotides. Although many studies have reported the cross-kingdom transfer between animals and parasites, plants and viruses (LaMonte et al., 2012; Feng and Chen, 2013; Buck et al., 2014; Han and Luan, 2015), in 2012, Zhang et al. demonstrated for the first time that osa-miR168a and other exogenous miRNAs abundant in rice plants are present in the sera and tissues of mice acquired orally through food intake. Functional studies in vitro and in vivo demonstrated that osa-miR168a binds the human/mouse low-density lipoprotein receptor adapter protein 1 (LDLRAP1) mRNA, inhibits the expression of protein in liver, and decreases the LDL removal from mouse plasma (Zhang et al., 2011). For the first time, Zhang and collaborators demonstrated that miRNAs contained in vegetal food regulate mRNA translation in a manner of mammalian functional miRNAs.
Although there are controversial opinions about the presence of small RNAs in tissues and the efficiency of dietary ingestion as the mechanism of cross-kingdom delivery (Dickinson et al., 2013; Snow et al., 2013), recently, it has been demonstrated that abundance in the serum of plant miR159 was inversely correlated with breast cancer incidence and progression in patients (Chin et al., 2016). As stated by the authors, these results demonstrate for the first time that a plant miRNA can inhibit cancer growth in mammals. Moreover, miRNA profiles of several plants with alimentary and medical interest have been sequenced to better investigate the sensible regulation process behind these molecules (Baldrich and San Segundo, 2016; Pirrò et al., 2016).
During the past years, several computational approaches such as miRanda (John et al., 2004), TargetScan (Lewis et al., 2005), PITA (Kertesz et al., 2007), and PicTar (Krek et al., 2005) have been elaborated to predict miRNA:mRNA interactions.
Most algorithms use experimental knowledge to develop a specific scoring system such as miRNA:mRNA partial complementarity, seed region, target position, and sequence conservation features. In 2013, Coronnello and Benos developed a web tool for combinatorial miRNA target prediction called COMIR, which combines four popular scoring schemes (miRanda, PITA, TargetScan, and mirSVR) to compute the potential of a gene to be targeted by a set of miRNAs (Coronnello and Benos, 2013).
In 2015, Shu presented comparative analysis and computational predictions of miRNAs that can be transferred into human circulation (Shu et al., 2015). However, no bioinformatic studies have been carried out to design, implement, and validate an algorithm to predict the action of plant miRNAs inside the mammalian host cell environment leading to cross-kingdom interactions.
For this reason, we rationalized the required sequence homologies between plant and mammalian miRNAs, suggesting for the first time the concept of cross-kingdom functional homology, and we developed MirCompare, a web application based on a strategy for the comparison between plant and mammalian miRNAs and their highly stringent selection to identify some possible plant miRNAs, with functional homologies with mammalian ones. Moreover, we conducted preliminary analysis to investigate potential human gene regulation by miR168a mimic from Moringa oleifera Lam. The reported analysis increases the information on plant miRNAs currently available and improves the knowledge on to the molecular mechanisms associated with nutritional and medicinal activities of this plant species.
2. Mircompare Development
According to Zhang et al. (2011), plant miRNAs in mammalian cells use the endogenous machinery to post-transcriptionally regulate their target genes.
Starting from this fundamental evidence, we propose a model where plant and mammalian miRNAs share the same gene targets if they show the following: (1) a sequence homology rate higher than a specific percentage and (2) a strict sequence homology related to the seed region.
MirCompare is based on an algorithm able to compare libraries of miRNAs belonging to organisms from plant and animal kingdoms, to find cross-kingdom functional homologies. As shown in Figure 1, the computational process is divided in two main phases—comparison and filtering.
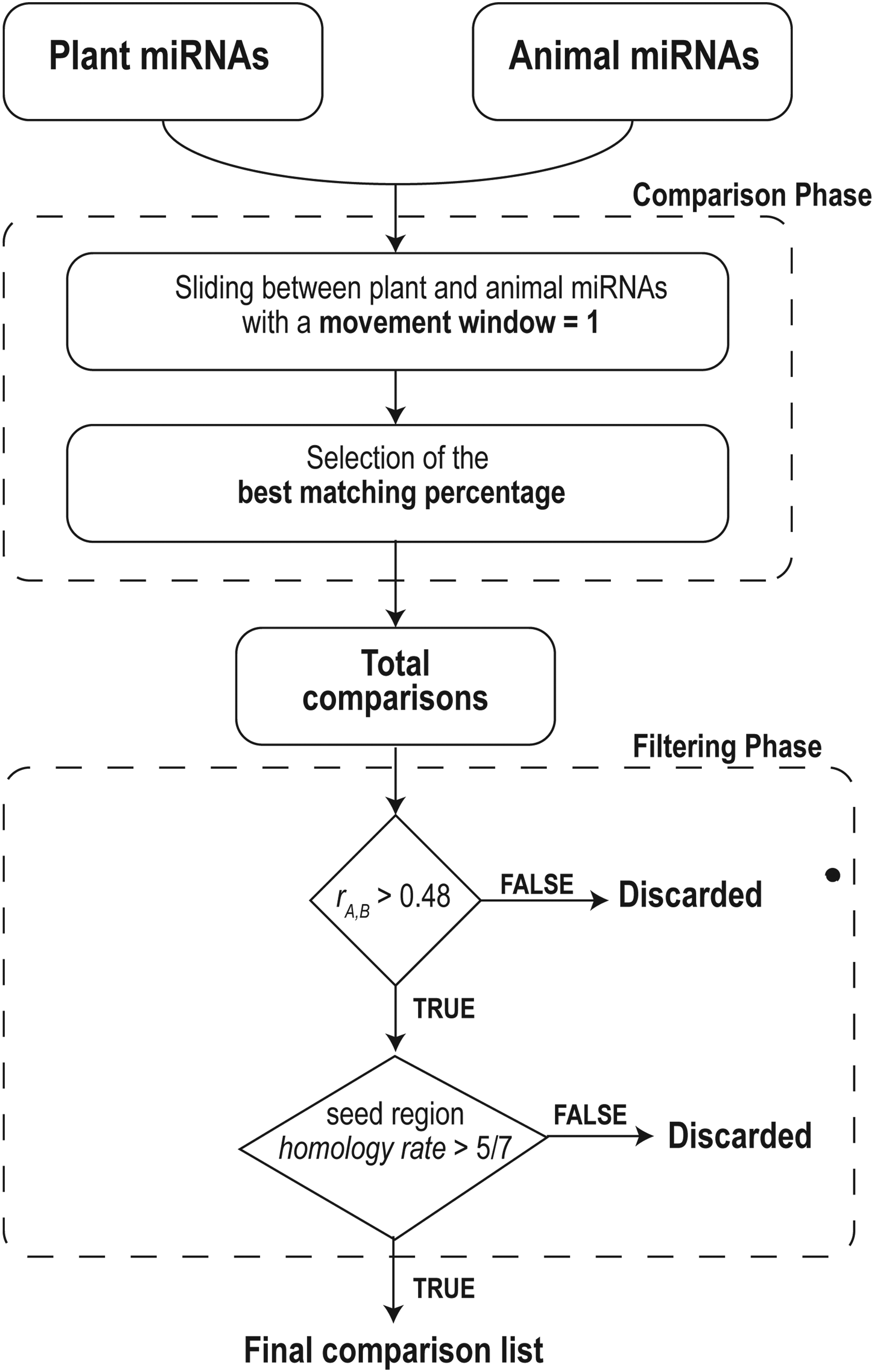
Workflow of the MirCompare algorithm. In the comparison phase, we evaluate the best alignment rate between each couple of miRNAs. In the filtering phase, we select only functional homologies using a double-step approach based on the evaluation of the overall homology and the seed region stringency.
2.1. Comparison phase
To evaluate the best alignment rate between each couple of miRNAs (A and B), the shorter sequence is sliding on the longer one, using a sliding window of 1 nucleotide, and for each step, an alignment score SA,B is calculated.
When all the possible alignment rates for each couple of uploaded miRNAs have been generated, the algorithm selects the best comparison rate (r):
The comparison phase computes a total of N × M different comparison rates, where N and M are, respectively, the length of the first and second miRNA data sets.
2.2. Filtering phase
In the filtering phase, the rules for the cross-kingdom functional sequence homology are applied. The process can be subdivided into two parts as follows. (1) Filter based on the overall sequence homology rate and (2) selection related to the seed region sequence homology.
To identify a cutoff for the best comparison rate between each miRNA couple (rA,B), 104 and 106 stochastic comparisons were generated, using two scramble sets of 102 and 103 sequences, respectively (Fig. 2A, B). According to Wang et al. (Wang, 2013), base composition of mammalian miRNAs show an evolutionary conservation and are GU rich. For this reason, we decided to build the sets of random sequences according to the experimental evidences. More in detail, each set is composed of sequences of length between 17 and 25 nucleotides with a percentage of A and C of 20% and G and U of 30%. To establish the minimum r-value threshold able to produce a statistically significant alignment between a couple of miRNA sequences (if the comparison between a couple of miRNAs is better than we would expect between any two random sequences), we plotted the r-value frequency distribution in above described stochastic comparison analysis (Fig. 2A, B).
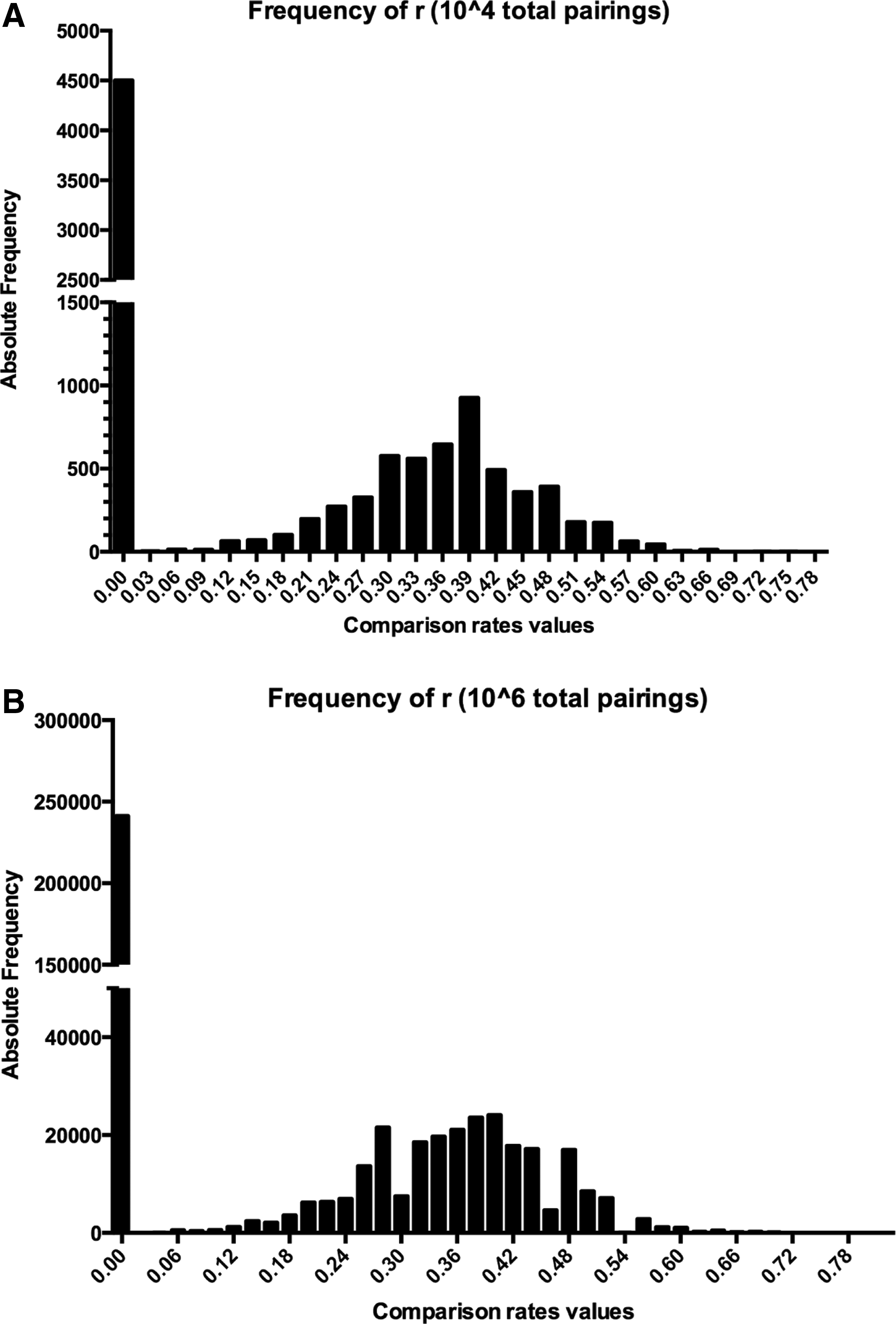
r-Value frequency distribution in two stochastic analysis involving 104
For each generated r-value, we calculated a p-value defined as follows:
In both the casual comparisons, a peak on 0 has been observed, confirming the casualty of the alignment. Looking at the r-value distributions, we also obtained similar statistically significant cutoffs. In the case of 104 comparisons, the lowest threshold value that generates a p-value ≤0.05, is 0.48, while in the case of 106, it is 0.46. For this reason, we decided to fix the r-value threshold at 0.48. MirCompare gives the possibility to arbitrarily change this value according to the desired stringency of the analysis.
Functional studies from Brennecke et al. (2005) have been conducted to identify the minimal requirements for a functional miRNA–target duplex in vivo. The magnitude of target regulation is not affected by a mismatch at positions 1, 9, or 10, but any mismatch from positions 2 to 8 causes a strong reduction. In addition, a minimal complementarity of four or five nucleotides into the seed region is necessary to confer target regulation. Therefore, we applied an additional filter, selecting only the comparisons that show a homology rate value higher than five out of seven nucleotides, into the seed region.
2.3. Computational analysis
2.3.1. Prediction of miRNA targets
To predict all the possible cross-kingdom targets in humans, we assumed that plant miRNAs regulate host mRNA translation in an analogous manner of mammalian functional miRNAs. To this end, the COMIR web tool (Coronnello and Benos, 2013) was used to rank human genes by their propensity to be regulated by human miRNAs showing functional homology with the plant ones. The COMIR output is provided as a list of “target gene–miRNA–COMIR score” elements.
2.3.2. Protein interaction analysis
The mentha interactome browser (Calderone et al., 2013) has been used to retrieve protein interaction network of the top predicted plant-regulated human genes and their first neighbor. Cytoscape software (Shannon et al., 2003) was used to manage the extracted network to filter relationships on the propensity of a couple of proteins to interact. The lowest threshold was set to 0.7.
2.3.3. Analysis on sequence conservation
To evaluate the conservation of miR168a in all sequenced plant species, Clustal 2.1 (Larkin et al., 2007) has been used.
2.4. Experimental validation of miRNA-mRNA cross-kingdom interaction
2.4.1. Transfection
We studied the biological activity of the synthetic mol-miR168a (UCGCUU GGUGCAGGUCGGGAC) that was transfected into the hepatocellular carcinoma cell line (HEPG2) by the Lipofectamine method (Hi-Fect; Qiagen) in accordance with the manufacturer's instruction (miRNA mimic and inhibitor experiment protocols; Qiagen) as already described (Pirrò et al., 2016).
2.4.2. Western blot analysis
Western blot analysis was performed in triplicate biological replication using the experimental procedure already described (Pirrò et al., 2016).
3. Results
3.1. Selection of M. oleifera miRNAs
In 2016, Pirrò and collaborators analyzed miRNA populations obtained from seeds of M. oleifera (Pirrò et al., 2016), with the aim of investigating a possible miRNA-mediated regulation process behind its medicinal factor. MirCompare has been used to identify M. oleifera miRNAs with functional homologies with human ones, comparing M. oleifera and Homo sapiens miRNomes.
As shown in Figure 3, the analysis of 98 M. oleifera (94 conserved and 4 novel) and 2042 H. sapiens miRNAs (www.mirbase.org) (Kozomara and Griffiths-Jones, 2010) generated a total of 285,880 different comparisons.

Schematization of MirCompare analysis between M. oleifera and H. sapiens microRNAs.
The exclusion of those with an r-value lower than 0.55, allowed to reduce the number to 904. After the second filtering phase over the seed region, only nine different comparisons have been obtained (Table 1) that involved eight M. oleifera and nine H. sapiens different miRNAs. As reported also in Pirrò et al. (2016), the mol-miR166i resulted in functional homologies with hsa-miR6503-3p that is involved in regulating inflammation (Ma et al., 2015), and mol-miR393c was homologous to hsa-miR548ah-5p that is involved in immune tolerance (Xing et al., 2015). Furthermore, mol-miR168a showed sequence homology with hsa-miR579, a human miRNA that normally regulates TNFα expression during endotoxin tolerance (El Gazzar and McCall, 2010).
miRNAs, microRNAs.
3.2. Computational prediction of human genes regulated by M. oleifera miRNAs
The COMIR web tool (Coronnello and Benos, 2013) was used to compute the potential of a human gene to be targeted by each M. oleifera miRNA highlighted by MirCompare (Table S2, Pirrò et al., 2016).
To better understand the putative role of exogenous miRNAs in the alteration of intracellular interaction networks, we selected the top 10 human genes for each M. oleifera miRNA (Table S2, Pirrò et al., 2016), and we built a protein interaction network (PPI) using the mentha interactome browser (Calderone et al., 2013). Mentha classifies interaction on type and method to assign a reliability score to each binary (protein A, protein B) interaction. The reliability score was used to filter the PPI network from the intrinsic noise of PPI information due to contradictory PPI information, inconsistent data curation, or curation errors. We set up a minimum score of 0.7 to filter interactions.
As described in Table 2, mol-miR168a regulates the expression of SIRT1, a hub protein with a key role in regulating cellular pathways such as apoptosis, cell cycle, and protein degradation. As shown in Figure 4 SIRT1 is a highly reliable direct interactor of P53 (Langley et al., 2002; Kim et al., 2007, 2008; Zhao et al., 2008; Inoue et al., 2011; Liu et al., 2011; Jain et al., 2012; Gonfloni et al., 2014). Moreover, SIRT1 is significantly elevated in human prostate cancer (Huffman et al., 2007), acute myeloid leukemia (Bradbury et al., 2005), and primary colon cancer (Stünkel et al., 2007). Based on these experimental evidences, we decided to focus our attention on mol-miR168a for further analysis.
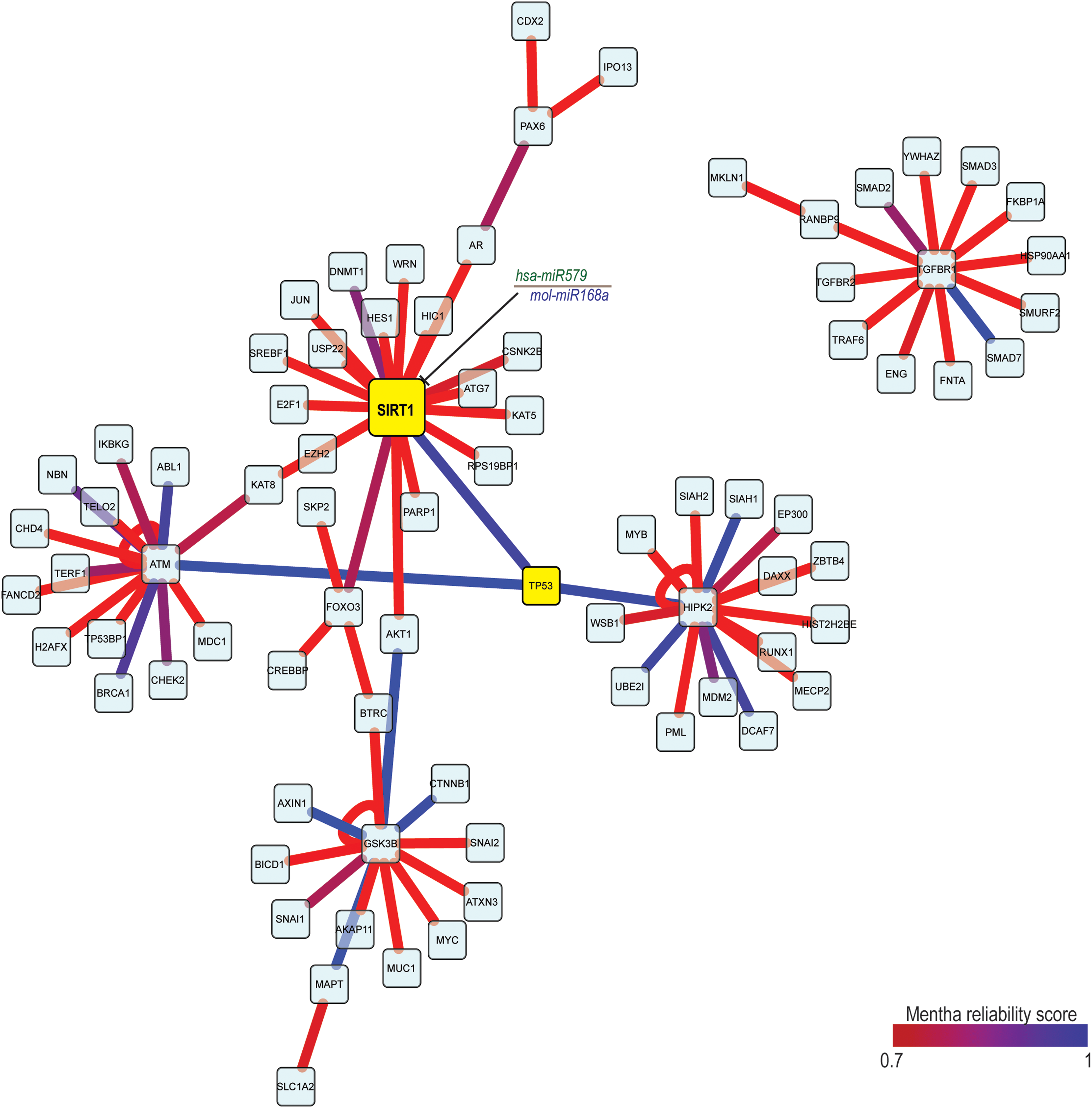
Protein interaction analysis for the genes showing best COMIR score.
COMIR, combinatorial miRNA target prediction.
3.3. Conservation analysis of plant miR168a family
As well described by Zhang and coworkers (Jones-Rhoades, 2011), an analysis on 481 miRNAs in 71 different plant species showed that miR168 family is found in at least 10 plant families and can be considered as highly conserved miRNA. In addition, high levels of bdi-miR168 and zma-miR168a have been identified inside human and porcine breast milk exosomes, respectively (Lukasik and Zielenkiewicz, 2014). To understand how small variations in the miRNA sequence could affect the action in human cells, we first evaluated the conservation rate of mol-miR168a in 18 plant organisms stored inside miRBase (Kozomara and Griffiths-Jones, 2010). Sequence alignment details (Fig. 5A) and the related dendrogram (Fig. 5B) show the presence of three distinct clusters of sequences (Fig. 5C), which differ in one to two bases. The miR168a from Olea europaea showed discordance in 3′ and 5′ ends that may be related to a bias in the Illumina trimming process (Del Fabbro et al., 2013).
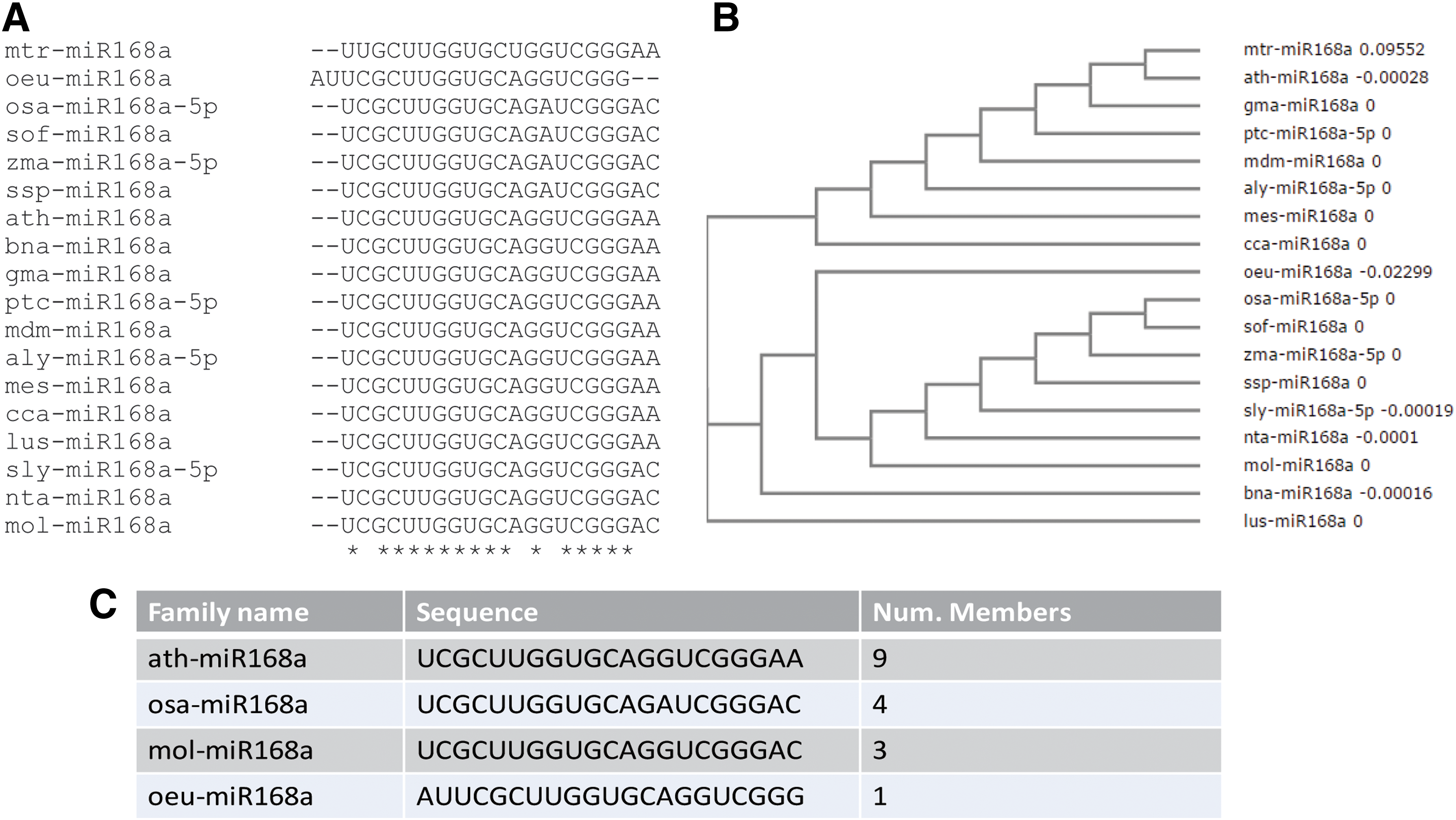
Analysis of sequence conservation for miR168a.
MirCompare was used to determine the relationship between conservation in sequence and functional homology with human miRNAs. As shown in Figure 6A, Arabidopsis thaliana, Oryza sativa, and M. oleifera groups share a total of 13 human miRNas that includes hsa-miR579. To study the dependency between miRNAs sequence conservation and action in human cells, independent COMIR (Coronnello and Benos, 2013) analysis on each plant miR168a was conducted. As clearly shown in Figure 6B, the three clusters of sequences share a total of 6244 genes (86.9%), and SIRT1 is included in this group.
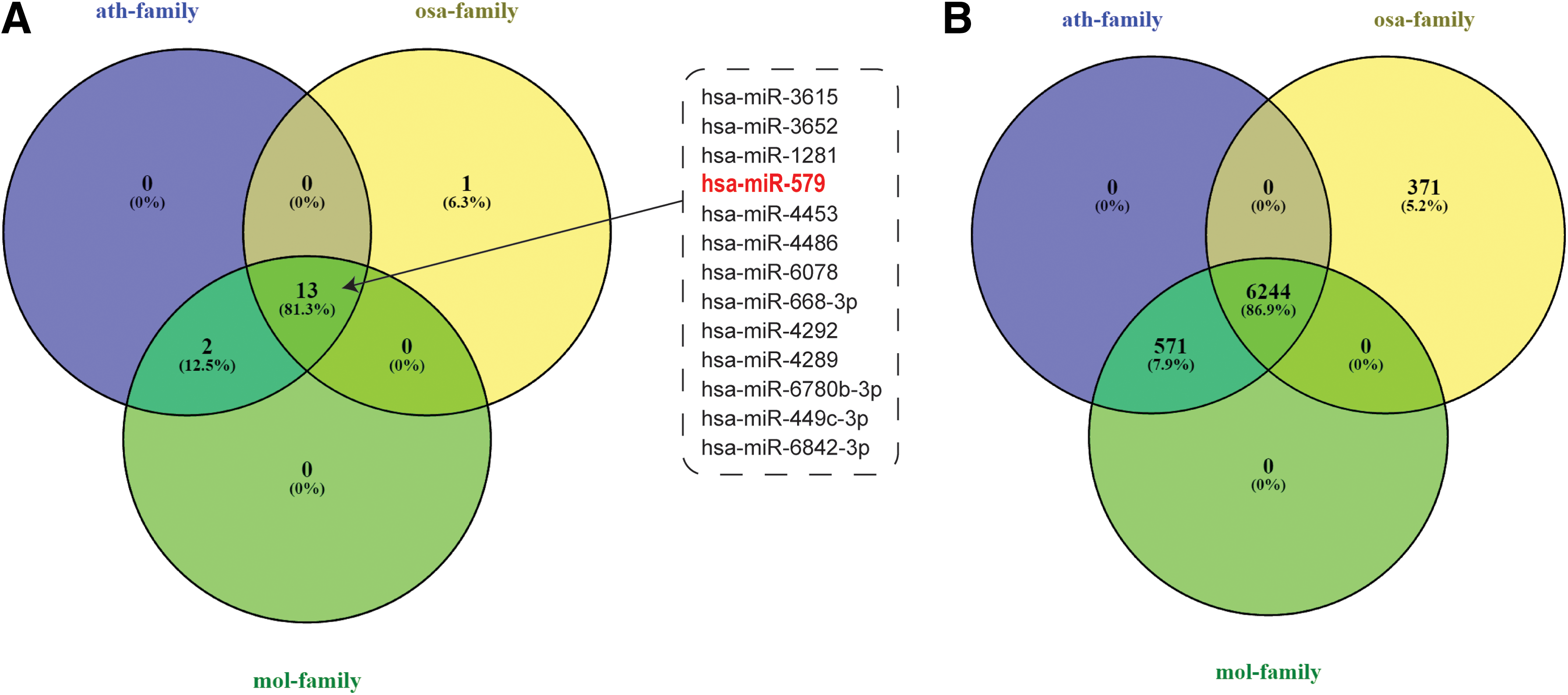
Overlapping rate of putative human functional homologs
3.4. Transfection efficiency and protein modulation
The formal evidence that mol-miR168a identified by MirCompare inhibits the translation of SIRT1 when their synthetic mimics are transfected into the cancer cell line HEPG2 was previously demonstrated (Pirrò et al., 2016) and described here in Figure 7. As shown in Figure 7A, the efficiency of transfection has been assessed monitoring the increase of the percentage of fluorescent-positive (FL1-positive) cells by flow cytometry analysis. To confirm the effect of miRNA treatment, we have investigated the protein expression of SIRT1, a specific target of hsa-miR579 homologous of mol-miR168a. The transfection of mimics derived from plant miRNA sequences in HEPG2 cell determined a significant decrease of SIRT1 protein level in comparison with HF control samples (Fig. 7B, C).

. Experimental validation of miRNA-mRNA human gene regulation.
4. Discussion
The possibility to identify vegetal miRNAs able to bind and regulate the human genome expression will be of paramount importance for better knowledge of nutritional value of our foods.
From the studies of Zhang's group that first demonstrated the presence of O. sativa miRNAs in the sera and tissues of mice, the scientific community has much debated to better elucidate this discovery. In this context, functional miRNA studies demonstrated that ICR mice fed with plant total RNA in quantities of 10–50 μg showed miR172 from Brassica oleracea as the most abundant exogenous miRNA in the sera, feces, and tissues in intestine, stomach, spleen, liver, and kidney (Liang et al., 2014). Encouraged by these discoveries, we shaped the concept of cross-kingdom functional homology, developing a completely new algorithm for the comparison of vegetal and mammalian miRNAs and implementing MirCompare, a free, fast, and user-friendly web application.
Given a collection of vegetal and plant miRNAs, MirCompare applies a double-layer filtering approach, respectively, based on overall and seed region homology rates.
Computational analysis on two different collections of stochastic sequences (102 and 103, respectively) showed that an r-value cutoff of 0.48 is required in the first-layer filtering, to extract statistically significant comparisons. This means that only 48% of overall homology between plant and mammalian miRNAs can lead to a functional correlation, emphasizing the concept of “functional homology.” According to the functional studies conducted by Brennecke et al., MirCompare further improves the analysis quality, applying a second-layer filter that takes into account only the comparisons, with strict homology related to seed region.
As study cases, M. oleifera and H. sapiens miRNomes have been provided to MirCompare to select a strict number of plant miRNA members with a potential genomic regulative role into human cells.
M. oleifera is one of the best known, most widely distributed, and most useful nutritional and medicinal plants (Anwar et al., 2006; Shahzad et al., 2013; Abdull Razis et al., 2014). The leaves are a source of natural antioxidants (Asma et al., 2005), vitamins, minerals, proteins, and essential amino acids (Anwar et al., 2006; Abdull Razis et al., 2014). A recent collaboration between universities in Italy and Cameroon has investigated the antioxidant and antitumor properties of M. oleifera (Canini, 2013).
Using low cutoff values for both the filtering layers, MirCompare highlighted 8 putative plant miRNAs with cross-kingdom potential, starting from 285,880 possible comparisons. Bioinformatic predictions of all the possible targets into human cell system for M. oleifera miRNAs highlighted a putative role of mol-miR168a in the active regulation of SIRT1. Protein network analysis highlighted the central role of SIRT1 in the regulation of P53 as a transcription factor. As well described by Gonfloni and collaborators, both SIRT1 and P53 regulate metabolism, stress signaling, cell survival, cell cycle control, and genome stability (Gonfloni et al., 2014).
Sequence analysis of miR168a sequences in 18 different plants show the presence of three different subfamilies that differ in one to two nucleotides. The synergic use of MirCompare and COMIR clarifies that small variations in miR168a sequences do not significantly affect homologies with human miRNAs (13) and their putative targets in humans (6244). Transfection experiments conducted by Pirrò et al. (2016) provided the formal evidence that mol-miR168a identified by MirCompare inhibits the translation of SIRT1, when their synthetic mimics are transfected into the cancer cell line HEPG2.
Further analysis will be carried out to better investigate the molecular mechanisms underlying the cross-kingdom interactions highlighted by MirCompare. Additional molecular and pharmaceutical approaches need to validate the clinical utilization of plant miRNAs in the treatment of cancer and other dysregulatory diseases. The ease of use of our web application, coupled with its high calculation speed, and flexibility in the selection of analysis parameters may help and allow the scientific community to better focus their attention on the cross-kingdom regulation mediated by plants and all the possible implications in the treatment of several diseases. This article mainly focuses the attention on the concept that cross-kingdom regulation of human genome is not a peculiar funding, as suggested by Zhang et al. in 2012, but a more generalized phenomenon linked to the historical Greek medical concept that “we are what we eat.”
Footnotes
Acknowledgments
The authors thank Dr. Alberto Calderone, Institute for Systems Analysis and Computer Science “Antonio Ruberti”—National Research Council, Professor Massimo Amicosante, Department of Biomedicine and Prevention, University of Rome “Tor Vergata,” Dr. Marco Cirilli and Professor Rosario Muleo, Department of Science and Technology for Agriculture, Forestry, Nature, and Energy, University “La Tuscia,” Viterbo for language revision and helpful discussion.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.