
Abstract
Abstract
RNA sequencing (RNA-seq) has emerged as the method of choice for measuring the expression of RNAs in a given cell population. In most RNA-seq technologies, sequencing the full length of RNA molecules requires fragmentation into smaller pieces. Unfortunately, the issue of nonuniform sequencing coverage across a genomic feature has been a concern in RNA-seq and is attributed to biases for certain fragments in RNA-seq library preparation and sequencing. To investigate the expected coverage obtained from fragmentation, we develop a simple fragmentation model that is independent of bias from the experimental method and is not specific to the transcript sequence. Essentially, we enumerate all configurations for maximal placement of a given fragment length, F, on transcript length, T, to represent every possible fragmentation pattern, from which we compute the expected coverage profile across a transcript. We extend this model to incorporate general empirical attributes such as read length, fragment length distribution, and number of molecules of the transcript. We further introduce the fragment starting-point, fragment coverage, and read coverage profiles. We find that the expected profiles are not uniform and that factors such as fragment length to transcript length ratio, read length to fragment length ratio, fragment length distribution, and number of molecules influence the variability of coverage across a transcript. Finally, we explore a potential application of the model where, with simulations, we show that it is possible to correctly estimate the transcript copy number for any transcript in the RNA-seq experiment.
1. Introduction
I
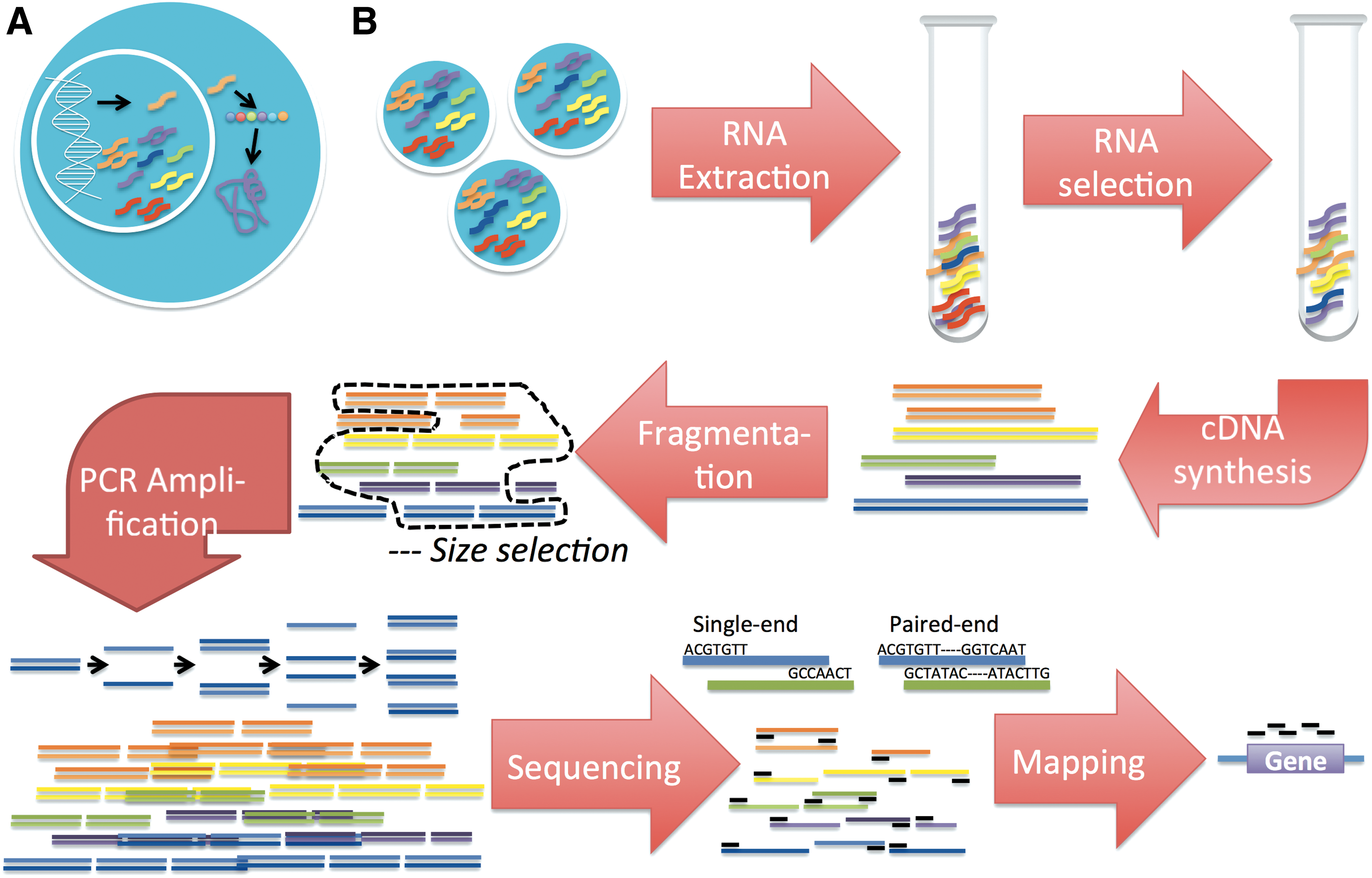
Recent RNA-seq projects have been directed toward measuring lower amounts of starting RNA (Adiconis et al., 2011; Saliba et al., 2014), where the majority of the fragments from each molecule are sampled and thus fragment starting-points (SPs) (boxes shaded red in Fig. 2) on a transcript are not independent. Here, we suggest a model of unbiased fragmentation of a single transcript in which we enumerate all possible ways to obtain the maximum number of fragments of a desired length from a transcript. Under our model, we produce the expected coverage for varying fragment length to transcript length ratios. To inch closer to reality, we also explore the influence of multiple fragment lengths and its distribution on the coverage profile. We simulate unbiased sequencing experiments that start with a fragmentation process based on our model and investigate the possibility to estimate the number of molecules fragmented in the RNA-seq experiment through SP distributions.

The fragment placement pattern space for various transcript lengths.
2. The Fragmentation Model
2.1. Enumerating the fragmentation pattern space for a single transcript
We propose a simple model of fragmentation: A fragmented molecule is represented with fragments of length, F, assigned on nonoverlapping positions of a transcript of length, T (see Table 1 for notations). Fragments may be assigned with gaps between them but gap lengths must be shorter than a fragment length—that is, we assume that fragmentation of a molecule involves exhaustive fragment placement. We define each unique configuration of exhaustive fragment placement as a Fragmentation Pattern and the Pattern Space comprises all unique fragmentation patterns possible, given F and T (some examples shown in Fig. 2).
The observation that fragmentation patterns of shorter transcripts reoccur within longer transcripts (Fig. 2, bordered orange boxes) naturally led us to a recursion to compute the pattern space. In exhaustive fragmentation, the left-most fragment placed on a transcript may not be more than F base-pairs (bp) away from the start of the transcript. As such, the fragmentation patterns of the pattern space can be divided into F parts, where fragmentation patterns of each part have the left-most fragment beginning on the same distinct position and the pattern space for a shorter transcript on the remaining length. The number of fragmentation patterns (FP) in the pattern space for a given T and F, is the sum over the F parts, of the number of unique fragmentation patterns that exist in the pattern space for the remaining length of the transcript, after placement of the first fragment:
where
We assume that in unbiased fragmentation, every fragmentation pattern is equally likely. Therefore, for each position along the transcript, we sum the coverage contributed from the fragments observed in the pattern space to obtain the expected coverage profile (
The general formula for the coverage profile 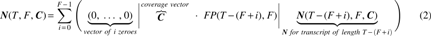
where
The coverage vector
This allows one to generate any of the desired profiles, that is,
2.2. Fragment length to transcript length ratios influence coverage profiles
Figure 3 shows the

Fragment length to transcript length ratios influence variability of coverage along the transcript. The expected SPPs
2.3. Read length to fragment length ratios influence the read coverage profile
When we distinguished R from F in our model, we find that the smaller the ratio of R to F, the more pronounced the differences between peaks and valleys of the

The expected read coverage profiles obtained from the enumerated pattern space for placement of fragments of length 200 bases long on a transcript of 1000 bases long and where the read lengths are 50, 75, 100, and 150 bases long.
2.4. Towards more realistic assumptions
2.4.1. Including a range of fragment lengths
In the experimental fragmentation process, a range of fragment lengths are produced. We, therefore, updated the model to enumerate all unique ways to place a range of fragment lengths,
where
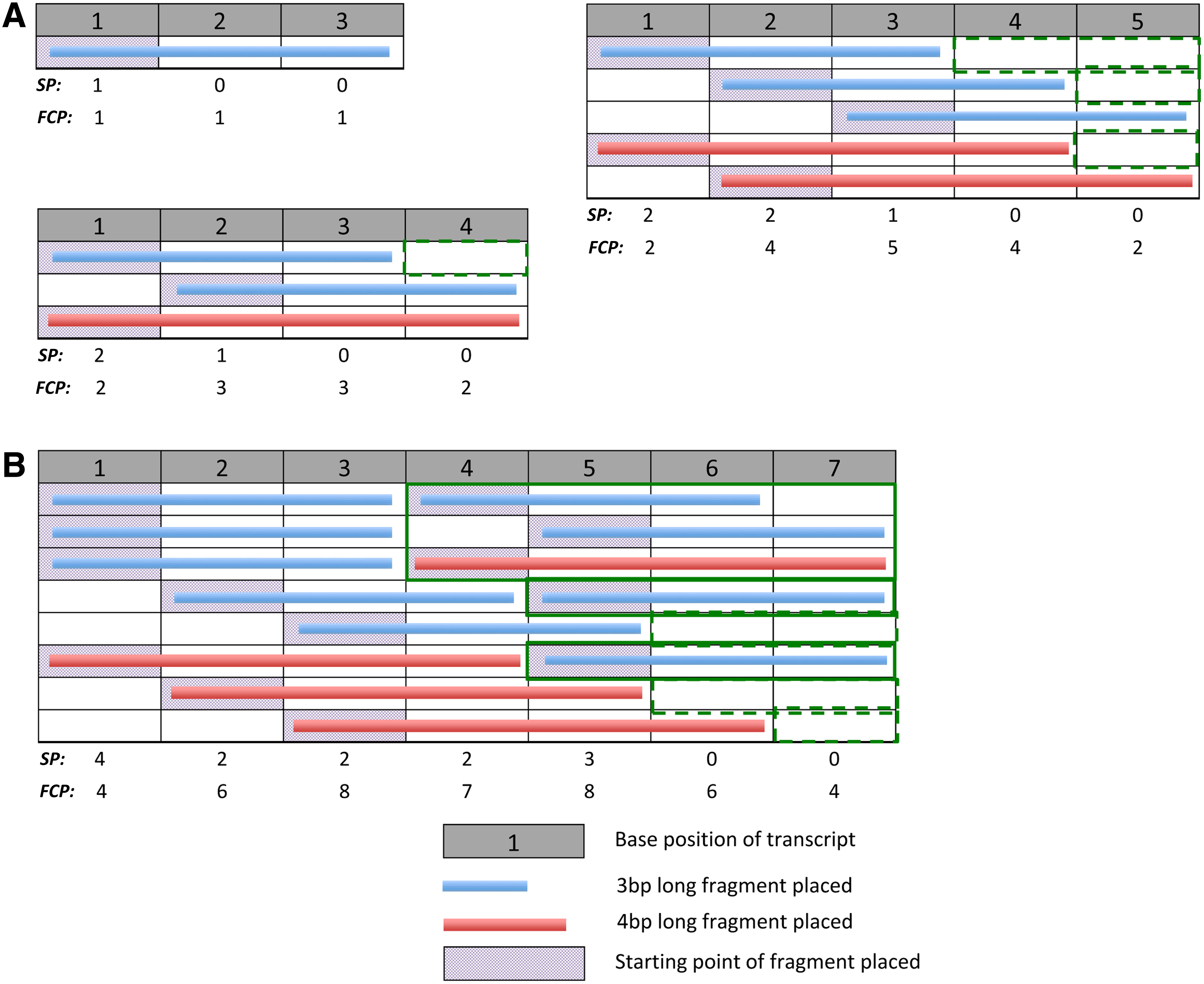
The fragment placement pattern space for various transcript lengths.
The general formula for the coverage profile, 
where
and
2.4.2. Imposing an experimentally derived fragment length distribution on the pattern space
In an RNA-seq experiment, fragment lengths occur at different frequencies. The fragment length distribution can be measured before sequencing of the input DNA (Panaro et al., 2000), but this information is typically not provided with RNA-seq data sets. We downloaded an RNA-seq library (SRR897347) of synthetic sequences (ERCC) from the Sequencing Quality Control (SEQC) project (SEQC-Consortium, 2014) and mapped the reads to a reference of ERCC sequences (Rosenbloom et al., 2013) with NextGenMap (Sedlazeck et al., 2013) using parameters—min_identity 0.9 and min_residues 1—such that the full length of read is mapped (note that 18.57% of reads remained unmapped). An empirical distribution of fragment lengths was determined from the difference between genomic positions of two extreme ends of mapped read pairs for the sequence ERCC-00002. We used this empirical distribution to linearly interpolate unobserved fragment lengths within the range of observed fragment lengths using na.approx (Zeileis and Grothendieck, 2005) in R. Next, the weight,
W(Fj): Weight for fragment length Fj
We modify Equation 4 to obtain the general formula, 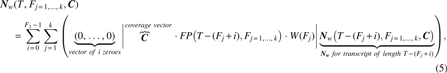
where
2.5. Fragment length range influences the coverage profile
We compared the results of enumerating the pattern space using a single fragment length (151 bp—the median fragment length mapped to ERCC-00002) to the results of enumerating the pattern space using a range of fragment lengths (100–960 bp—the range for fragment lengths mapped to ERCC-00002) with T = 1000 bp with R = 100 bp. The
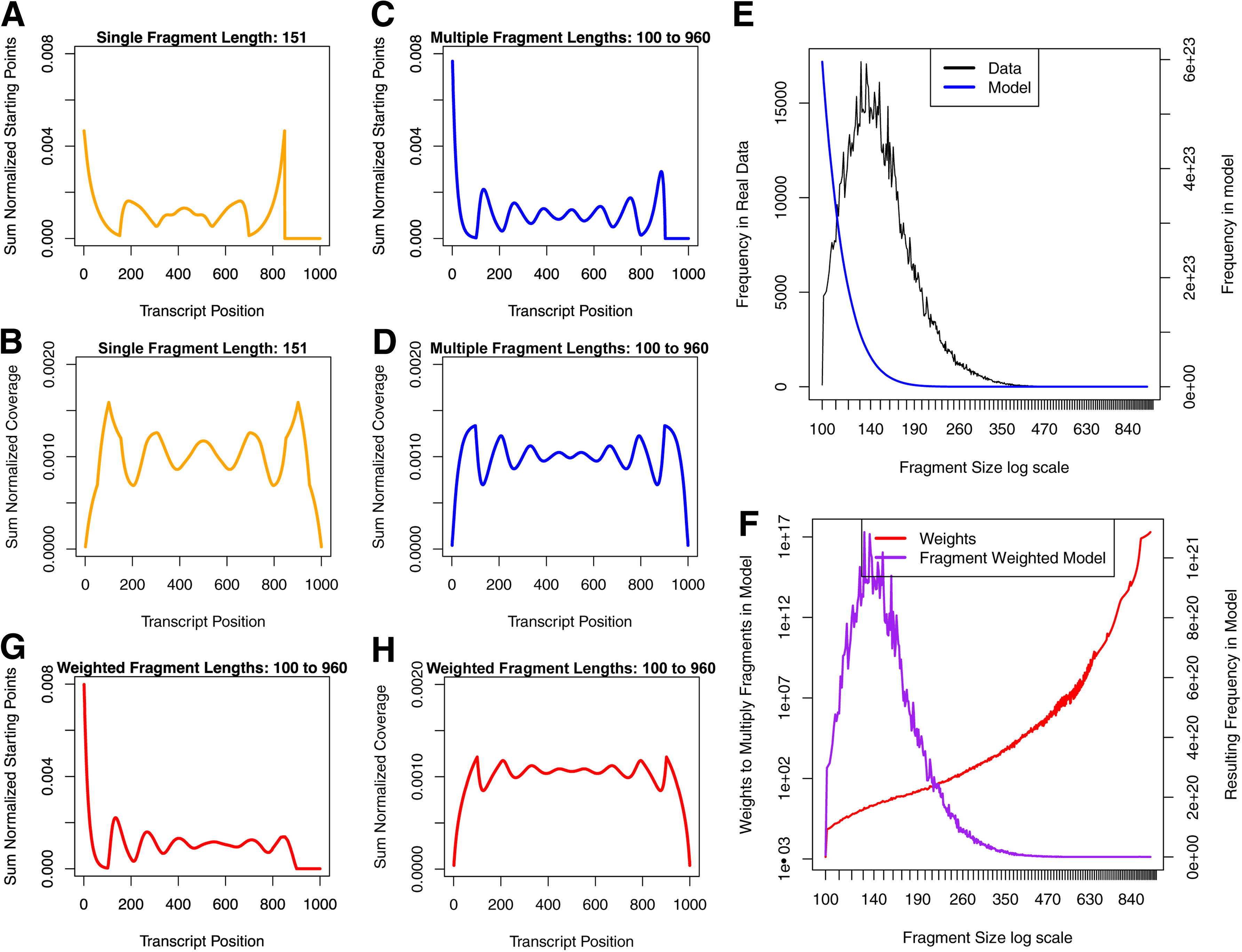
The influence of fragment size range and distribution on the
2.6. Fragment length distribution influences the coverage profile
The fragment length distribution from the enumerated pattern space is L shaped, whereas the empirical fragment length distribution that was inferred from reads mapping to the sequence ERCC-00002 is bell shaped (Fig. 6E). Imposing the empirical fragment length distribution on the pattern space using weights (Fig. 6F) results in less pronounced differences between peaks and valleys in the second half of the
2.7. Simulating the RNA-Seq experiment: sampling of the pattern space
FP for a specific fragment length grows exponentially with increasing transcript length (Fig. 7A). As the number of copies of a transcript is less than the number required to represent the full pattern space, we “fragmented” a defined number of molecules by exhaustively assigning fragments on the transcript based on the
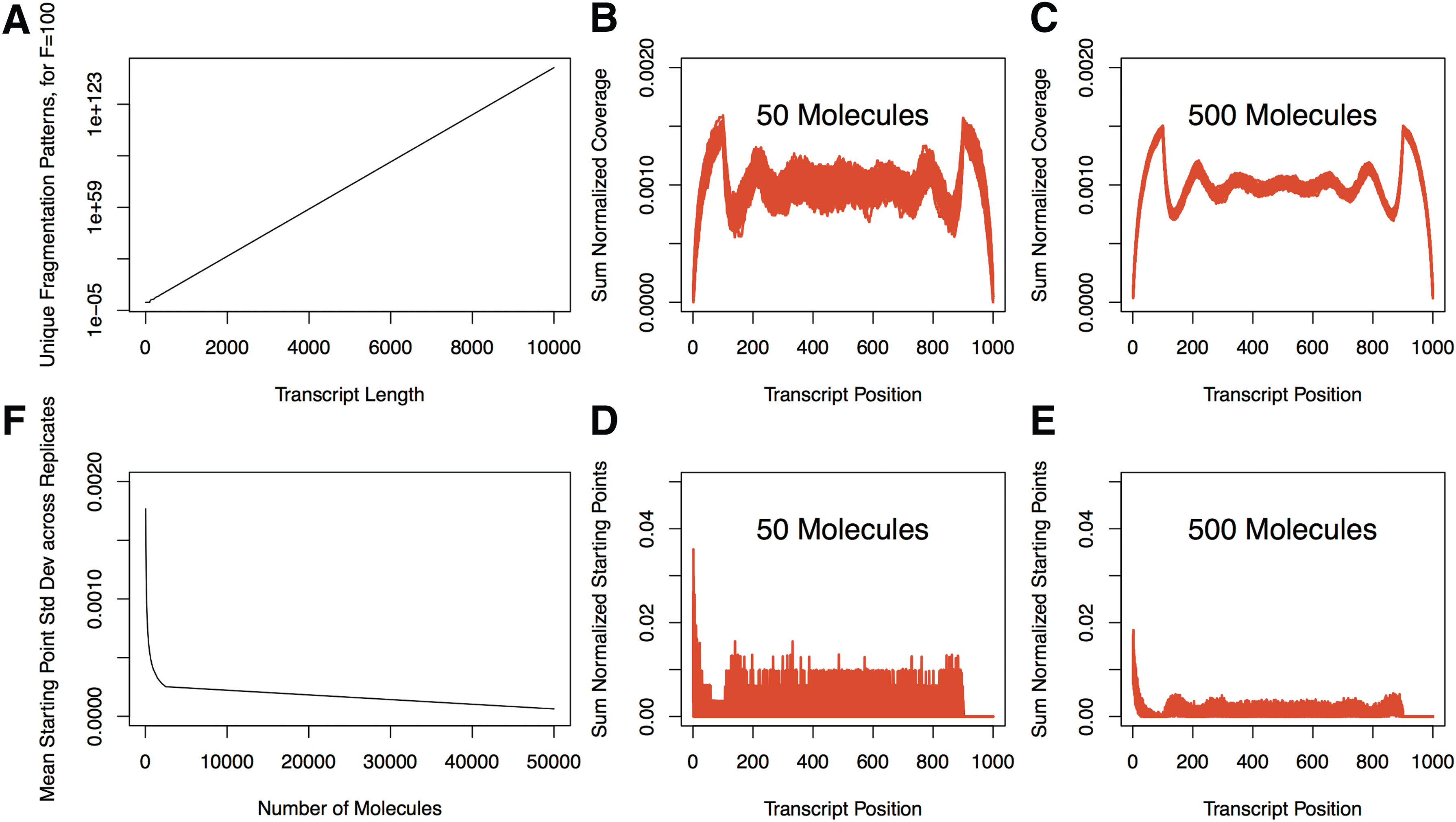
Sampling from the pattern space.
2.8. Estimating the original number of molecules
With the same procedure described in the previous section we generated an additional test profile for each of the 43 values evaluated between 50 molecules and 50,000 molecules and compared the test profiles with all 43 sets of the 100 repetitions. To make comparisons between profiles, the
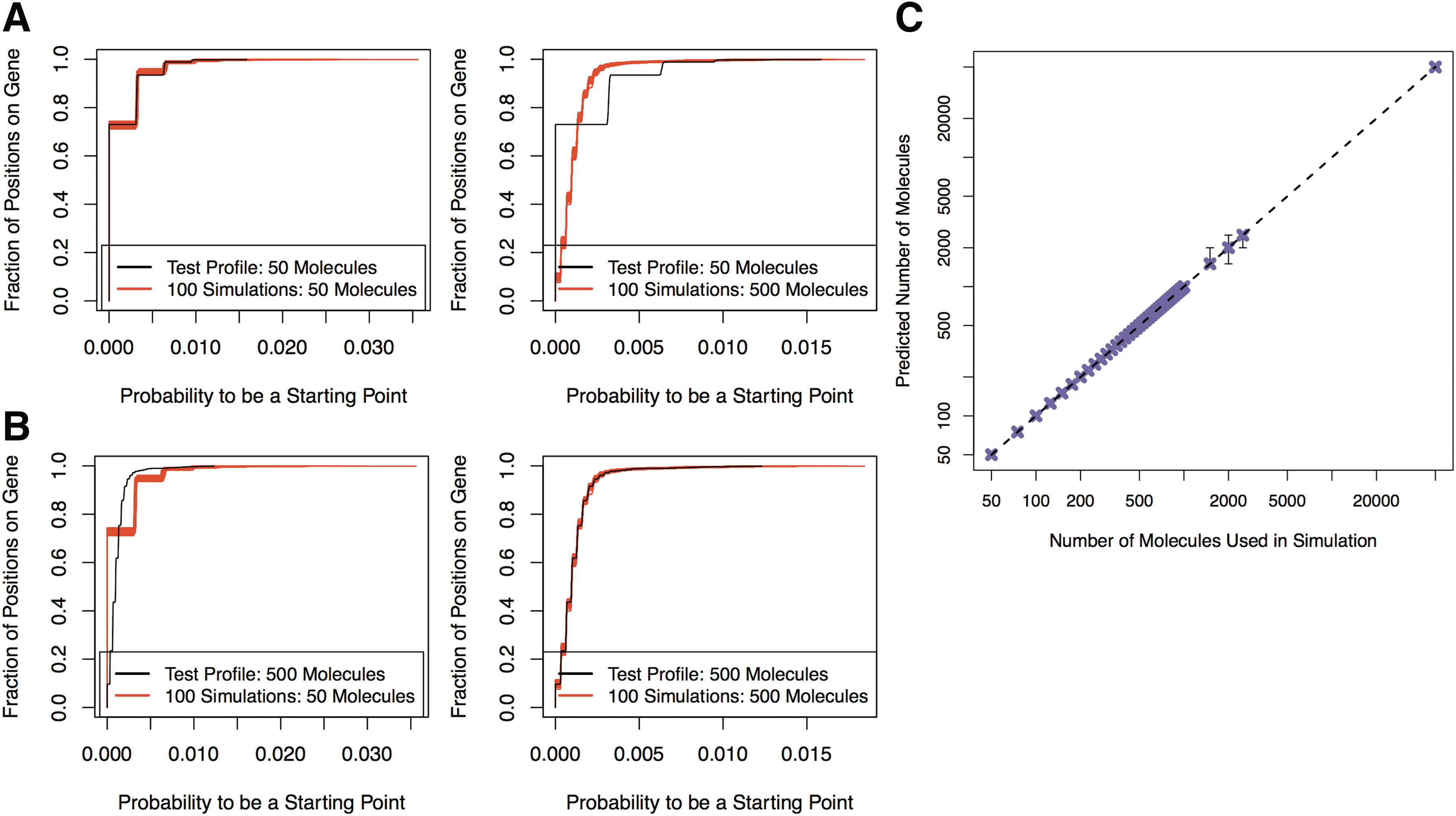
Estimating the number of molecules that were sequenced.
The distance between two ECDFs,
where
Based on the set with the smallest distance to the test profile ECDF, we predicted the original number of molecules the test profile was generated with. We found that the
3. Discussion
This article explored a model for the expected distribution of fragment SPs for unbiased coverage across a single transcript in RNA-seq. We model the products of fragmentation by enumerating all possible unique ways to obtain the maximum number of fragments of a desired length from a transcript. This is pertinent to recent RNA-seq projects of smaller amounts of starting material (Adiconis et al., 2011; Saliba et al., 2014), where the majority of the fragments from each molecule are sampled. Read coverage in RNA-seq has so far been nonuniform; for instance, read counts modeled as Poisson variables with constant rates along each transcript fit data poorly (Li et al., 2010) and the majority of transcripts evaluated by Hower et al. (2012) were found not to have homogeneity of starting-point and fragment length distribution along the transcript. Although nonuniformity in coverage is often attributed to biases in the experiment (Wu et al., 2011; Benjamini and Speed, 2012), we found that the expected unbiased coverage profile is dependent on the fragment length to transcript length ratio, read length to fragment length ratio, and fragment length distribution. Our results have implications on the methods that assume the expected coverage is uniform (Jiang and Wong, 2009; Li et al., 2010; Hower et al., 2012). Based on our analysis, some variability in the SPs is expected and thus investigations of nonuniformity should be aimed at positions with observations outside these expected values.
As opposed to the Poisson (Jiang and Wong, 2009) and negative binomial (Miller et al., 2011) models that provide a numerical measure to capture the distribution of the data, the pattern space model is based on a spatial representation of the products of fragmentation. Identical configurations have also been described for a discrete car-parking problem (Texter, 1989), but unlike the car-parking problem, we assume all patterns have the same probability of occurring. We find this results in greater packing densities in the exhaustive fragmentation pattern space than the discrete car-parking problem (data not shown). In our analysis, we inferred the fragment length distribution of a transcript based on mapped reads, and this may not be the fragment length distribution after fragmentation. Nonetheless, the model provides insights into how the fragment length distribution influences the variability of the SP distribution.
In future, we plan to evaluate whether the trends we observed with our proposed pattern space model occur in experimental data sets that were prepared from both limited number of molecules and exhaustive sequencing. Given that we were able to accurately predict the number of molecules we used in RNA-seq simulations, it would also be of great interest to evaluate the potential to determine the original number of molecules in empirical RNA-seq data sets.
Footnotes
Acknowledgments
We thank Florian Pflug and Luis Felipe Paulin Paz for their helpful discussions and comments to the article. We also thank Stefanie Tauber and Maurits Evers for their earlier contributions. This work was supported by the Austrian Science Fund (FWF): Project W1207-809.
Author Disclosure Statement
No competing financial interests exist.