
Abstract
Abstract
The hydrophobic-polar (HP) model is commonly used for predicting protein folding structures and hydrophobic interactions. This study developed a particle swarm optimization (PSO)-based algorithm combined with local search algorithms; specifically, the high exploration PSO (HEPSO) algorithm (which can execute global search processes) was combined with three local search algorithms (hill-climbing algorithm, greedy algorithm, and Tabu table), yielding the proposed HE-L-PSO algorithm. By using 20 known protein structures, we evaluated the performance of the HE-L-PSO algorithm in predicting protein folding in the HP model. The proposed HE-L-PSO algorithm exhibited favorable performance in predicting both short and long amino acid sequences with high reproducibility and stability, compared with seven reported algorithms. The HE-L-PSO algorithm yielded optimal solutions for all predicted protein folding structures. All HE-L-PSO-predicted protein folding structures possessed a hydrophobic core that is similar to normal protein folding.
1. Introduction
A
Regarding ab initio methods, the hydrophobic-polar (HP) protein folding model (Dill, 1985) was proposed for predicting protein folding. In general, the HP model entails simulating the folding process of amino acid sequences on lattice models, such as triangular lattices (Bechini, 2013), and classifying all amino acids into hydrophobic (H) and polar (P) types. However, exploring the possibility of an extremely large folding process is necessary, and resolving a nondeterministic polynomial-time hard (NP-hard) problem for deriving optimal solutions remains a challenge (Crescenzi et al., 1998; Qian et al., 2007).
According to Anfinsen's thermodynamic hypothesis, an amino acid sequence may fold into a tertiary structure with a lower free energy level and may thus more closely resemble real protein folding (Anfinsen, 1973). This concept is also used for evaluation in HP model-based protein folding predictions. When two hydrophobic amino acids are adjacent to each other, a hydrophobic–hydrophobic (H–H) interaction may be generated. When the number of H–H interactions is increased, the predicted structure becomes more stable and highly similar to natural protein folding. Concerning two-dimensional (2D) lattice models, both square and triangular HP models have been proposed. A triangular lattice model was reported to have looser topological constraints, a higher number of simulated hydrophobic interactions, and higher efficiency in predicting protein folding, compared with a square lattice model (Bechini, 2013). Accordingly, the current study applied a triangular lattice model.
Because protein folding involves integration of the movement of each connecting amino acid, its prediction is a complex and time-consuming process even when the computation strategy is properly applied. Accordingly, using an algorithm with a convergence property to predict protein folding may prove helpful. Recently, a high exploration particle swarm optimization (HEPSO) algorithm was reported to perform effectively for convergence speed, global optimality, and solution accuracy (Mahmoodabadi et al., 2014). Therefore, we hypothesize that the HEPSO algorithm may improve the prediction of protein folding. However, the HEPSO algorithm may only focus on optimization for predicting protein folding because of its global search property. Recently, an NP-hard problem (Thilagavathi and Amudha, 2014) was reported to have been solved using local search methods such as the hill-climbing algorithm, greedy algorithm (DeRonne and Karypis, 2006), and Tabu table (Lin et al., 2014). Therefore, the combination of the HEPSO algorithm with local search algorithms for predicting protein folding warrants further investigation.
To improve the performance of protein folding predictions, we propose an HE-L-PSO algorithm that combines three local search algorithms with the HEPSO algorithm for providing higher quality solutions. In this study, we evaluated a simulation of protein folding on a triangular lattice by using three local search algorithms (hill-climbing algorithm, greedy algorithm, and Tabu table) as well as 20 amino acid sequences with known structures as examples.
2. Methods
We propose an HE-L-PSO algorithm that combines the HEPSO algorithm with the hill-climbing algorithm, greedy algorithm, and the Tabu table. The HEPSO algorithm can execute a global search in the search space, whereas the hill-climbing algorithm exchanges the contents of two rational structures in two particles and then attempts to exchange these two particles' information to determine a more effective structure. The greedy algorithm enhances prediction of protein structures by folding all amino acids in all directions to evaluate all fitness values and maintain optimal results. However, the greedy algorithm easily falls into a local optimal solution; thus, the Tabu table can store the current optimal solution through n iterations, and the contents of the particles are subsequently reset to prevent the same protein structure from being repeatedly predicted.
2.1. Particle swarm optimization
The particle swarm optimization (PSO) algorithm (Kennedy and Eberhart, 1995) is based on the foraging behavior of fish and birds. When the algorithm is executing a search process, each bird is regarded as a particle, and the birds seek food. For every period (iteration), all the birds share their experiences with other individuals. Each bird uses the shared information to improve the direction and speed of its search for food to increase the probability of finding food or to find higher quality food.
2.2. High exploration PSO
Mahmoodabadi et al. (2014) proposed the HEPSO algorithm that implements two methods for updating particles (Mahmoodabadi et al., 2014). The flowchart of the HEPSO algorithm is shown in the HEPSO part (without local search) of Figure 1. PB and PC affect the probability of using the artificial bee colony algorithm and genetic algorithm (GA), respectively, and the usage rate of the artificial bee colony algorithm is gradually reduced with the iteration; otherwise, probability of the GA and original PSO algorithm, which are used to update the probability of particles, would be increased.

Pseudocode of the HE-L-PSO algorithm. GA, genetic algorithm; HEPSO, high exploration particle swarm optimization; PSO, particle swarm optimization; TBP, HE-L-PSO.
2.2.1. Encoding
In the HP model prediction, each amino acid is treated as a dimension, and the content data represent the direction in which the next amino acid is folded. If the data value for a specific amino acid is 0, the next amino acid is folded to the upper left; if the value is 1, the amino acid on the right shifts to the upper right; if the value is 2, the next amino acid is to the right; if the value is 3, the next amino acid is to the lower right; if the value is 4, the next amino acid is to the lower left; and if the value is 5, the next amino acid is to the left. For example, if the data value for the third amino acid is 0, the fourth amino acid is to the upper left of the third amino acid.
2.2.2. Fitness calculation
In the HP model, the number of H–H interactions is regarded as an indicator of the stability of the algorithm in predicting protein structures. More interactions are expected to be closer to the natural folded state. Generally, fitness is represented by a negative value. The fitness of H–H interactions is counted for the two nearest hydrophobic amino acids. For example, the fitness values are −1, −4, and −2 for Figure 2A–C, respectively.
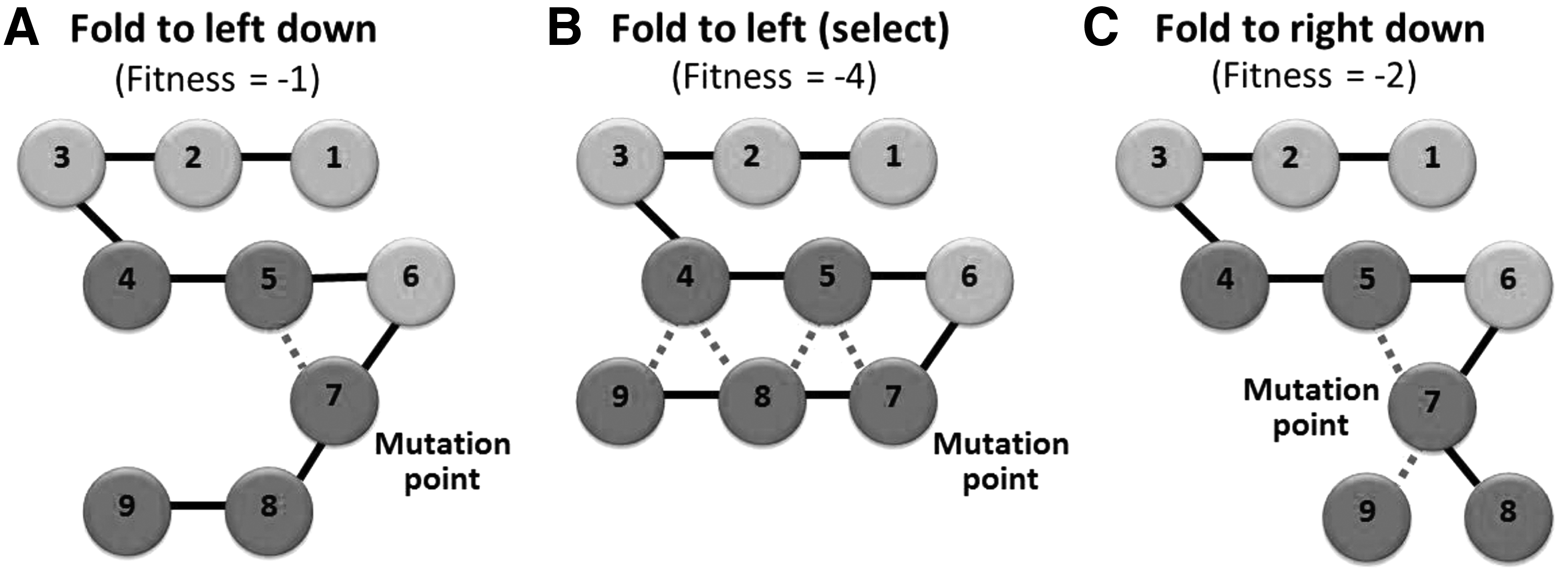
Example of protein folding prediction using the greedy algorithm. The seventh point is used as the example of the mutation point, and all possible folding directions are displayed, namely
2.2.3. Updating gbest and pbest
A local search algorithm is used after updating each particle in the HEPSO algorithm. The optimal solution for each particle (pbest) is updated before the end of the iteration. Specifically, the optimal solution for the particle in history is set to pbest. When the updating process is completed, the global best solution (gbest) is selected from all pbest solutions. Therefore, gbest and pbest guide the search directions for each particle in the next iteration of the HEPSO algorithm.
2.2.4. Updating particles
In the HEPSO algorithm, the optimal swarm experience (gbest) is used along with the previous optimal experience (pbest) to influence the next search direction (velocity), according to the following equations:
where v is the velocity, w is the weighting value, c1 and c2 are learning factors, and r1 and r2 are random numbers between 0 and 1; moreover, i represents the index of the particle among population, d represents the updated dimension, old represents the current iteration, new represents the newly calculated results (next iteration), pbest represents the optimal result of particles in previous iterations, and gbest represents the optimal result for all particles. Regarding calculation of the new velocity in Equation (1), the original velocity is first multiplied by the weighting value; subsequently, pbest and gbest are each subtracted from the contents of the original particle, and the corresponding results are multiplied by the random number and weight (r and c), after which the multiplication results are added to the product of the original velocity and the weighting value. In Equation (2), the contents of the original particle are updated by adding the new velocity, thus resulting in a new particle. The parameters are calculated according to Equations (3)–(7).
The values of c1 and c2 can be calculated using Equations (3) and (4), where i is the initial value of the learning factor, f is the final value of the learning factor, and t is the current iteration number. When the number of iterations increases, c1 becomes increasingly lower and c2 continues increasing. Therefore, the early iterations focus on a global search, whereas the later iterations focus on local search.
The value of w in Equation (1) can be obtained using Equations (5)–(7). Equation (7) can be used to calculate the value (d) for each particle. When the values for all the particles are calculated, Equation (6) can be used to obtain f, which is subsequently applied to Equation (5) to derive the weight values (w); the weighting values inherently range between 0.4 and 0.9. In Equation (7), Euclidean distance is applied to calculate the average distance between the current particle and other particles, where k is the dimension of each particle, D is the dimension of each particle size, i is the current particle, and N is the total number of particles. For all calculated di values, the optimal fitness of particles is considered to be dg. Moreover, dmax and dmin are the maximum and minimum values of all calculated di, respectively.
Equation (8) presents the artificial bee colony algorithm, where old represents the current iteration, new represents the next iteration, i represents the current particle, d represents the dimension to be calculated, x represents the particle itself, and r represents a random number between 0 and 1.
Equation (9) is based on the GA concept, where old denotes the current iteration; new denotes the next iteration; pbest denotes the optimal result of particles in previous iterations; gbest denotes the optimal results for all iterations; c2 denotes a learning factor, which is calculated according to Equation (4); v denotes the new speed, which is used in Equation (2) to update the contents of the particle; and i and d denote current particles and the dimension.
2.2.5. Local search
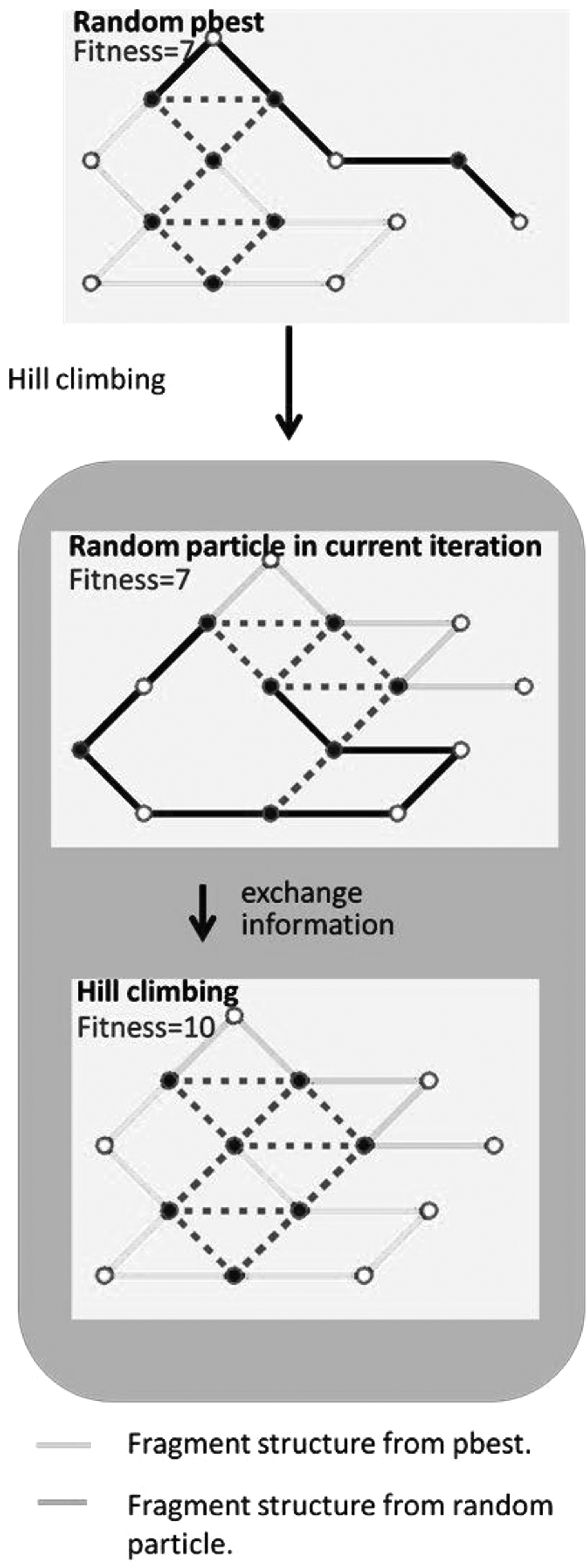
(1) Hill climbing. The hill-climbing algorithm (information exchange) is applied to derive two predictions of protein structures. If the updated prediction is superior to the original, the original information is replaced. The pseudocode of the hill-climbing algorithm is presented in Figure 1 (center). This algorithm (Lim et al., 2006) entails using current information to continue searching for superior results. In this study, we used the exchange information to design the climbing algorithm. As shown in Figure 3, the particle with the optimal solution in previous iterations (pbest) is randomly selected for information exchange with other random current particles. Information exchange is performed through a two-point crossover process. Once two random numbers are selected, all resource information between these two selected dimensions is exchanged. For example, when dimensions 3 and 6 are selected, all information from dimensions 3 and 6 is exchanged to obtain two new results.
(2) Greedy algorithm. If the hill-climbing algorithm produces two structures that are inferior to the original, the greedy algorithm can be used to strengthen the predictions. The pseudocode of the greedy algorithm is shown in Figure 1 (center). The greedy algorithm (DeVore and Temlyakov, 1996) is currently used to select the most favorable situation that is suitable for the exchange equivalent of the minimum coin problem. In this study, the amino acid sequence was designed to fold in all directions. Finally, only the optimal predictions of structures are chosen. As illustrated in Figure 2, the number presented in each circle is the order of the amino acid sequence. The dotted line represents H–H interactions; more dotted lines represent higher stability in predicting protein structures, which is closer to the natural folded state of proteins. In this study, we assumed that the seventh amino acids are mutated; therefore, we attempted to simulate the folding of this dimension in various directions such as left down (Fig. 2A), left (Fig. 2B), and right down (Fig. 2C). Among the predicted structures, the optimal structure with the highest H–H interactions is used to replace the original information. The fitness calculation is described in the following sections.
(3) Tabu table. The hill-climbing and greedy algorithms are executed several times, after which pbest and gbest are updated. When a certain set of iterations is reached, the optimal structure can be stored in the Tabu table, and the contents of all particles are reset. Future prediction processes are designed such that prediction of the same or similar structures is avoided. The pseudocode of the HE-L-PSO algorithm is displayed in Figure 1 (lower part).

Example of information exchange in the hill-climbing algorithm. The information between dimensions 3 and 6 is exchanged for pbestrand and particlerand, thus yielding solutionnew1 and solutionnew2, respectively.
The Tabu table was proposed by Glover (1989). The Tabu table records short-term results to prevent duplicate search processes in algorithms. This feature can prevent some degree of the optimal solution search process being trapped into a local optimum solutions. Moreover, the feature can effectively compensate for the shortcomings of the greedy algorithm in this study because we can record optimal results after every few iterations and then reset the contents of all particles, thus preventing the previously searched direction from being searched again.
2.3. Parameter settings
The parameter settings of the HE-L-PSO algorithm are described as follows. In this algorithm, PA represents the probability of particles [Eq. (8)], which is updated by the GA; the value of PA was set to 0.05 in this study. In addition, PB represents the probability of particles [Eq. (9)], which is updated by the artificial bee colony algorithm; in this study, the value of PB was set to 0.95. The numbers of particles and iterations for the PSO algorithm were set to 100 and 200, respectively, where c1 and c2 represent learning factors and i and f represent the initial and final values of the learning factors, respectively. Furthermore, C1i, C1f, C2i, and C2f were set to 2.5, 0.5, 0.5, and 2.5, respectively. The update speeds Vmin and Vmax were set to −5 and 5, respectively, because the triangular lattice model used for the prediction involved only six directions. In addition, the minimum and maximum search ranges were set to 0 and 5, respectively. The number of local search processes by the hill-climbing and greedy algorithms was set to 64. The Tabu table (reset in the iteration) was set to save gbest and reset the information after 20 iterations.
3. Results
3.1. Data set
As presented in Table 1, 20 sequences were used to predict the protein structure. The accuracy of prediction of the 2D triangular lattice model was evaluated and compared with a known optimal structure (Krasnogor and Smith, 2000). Regarding the contents of sequences, H and P indicate hydrophobic and polar amino acids. The prediction process typically assigns a folding direction for each amino acid, and the next amino acid is thus folded in that direction. The first amino acid does not affect the folding structure in any direction, and the last amino acid does not have a next object. Therefore, in the encoding process, the number of dimensions for each particle is equal to the length of the amino acid sequence (−2).
H, hydrophobic; P, polarity.
3.2. Comparison of optimal prediction results among several algorithms
The prediction performance of the multimeme memetic algorithm (MMA) (Krasnogor et al., 2002) for the 20 sequences (Table 1) is presented in Table 2. We compared optimal prediction results of the proposed HE-L-PSO algorithm with those of the MMA algorithm. The MMA algorithm did not predict the first sequence. When the amino acid sequence length was equal or greater than 24, the MMA algorithm could not determine the optimal structure.
Sequences from Table 1.
The optimal values are reported in the literature (Krasnogor and Smith, 2000). Values listed in this table represent optimal prediction results from different algorithms in 25 experiments.
The three local search algorithms comprise the hill-climbing algorithm, greedy algorithm, and Tabu table.
HE-L-PSO is the combination of the HEPSO algorithm and three local search algorithms. HE-L-PSO is visualized in Figure 6.
HEPSO, high exploration particle swarm optimization; MMA, multimeme memetic algorithm (Krasnogor et al., 2002); ND, not shown in the literature.
Notably, the HEPSO algorithm alone was ineffective in predicting most of the 20 sequences. Three local search algorithms alone demonstrated more effective performance in protein folding prediction than did the HEPSO algorithm alone; nevertheless, the local search algorithms alone still did not attain optimal performance for sequences 18–20. By contrast, the proposed HE-L-PSO algorithm, which combines the HEPSO algorithm with three local search algorithms, could execute optimal folding prediction for the 20 amino acid sequences (Table 2).
3.3. Success rate regarding optimal structure prediction
The number of optimal solutions derived by the HEPSO algorithm, three local search algorithms, and HE-L-PSO algorithm was recorded for comparison with other methods (GComa, SComa, GRand, SRand, simple memetic algorithm [SMA], and GA) (Smith, 2005) by testing each sequence 25 times (Table 3). The optimal performance regarding folding prediction was defined as 100% reproducibility (25/25). For the shortest sequence (i.e., sequence 1), three methods (SComa, SMA, and HE-L-PSO) could constantly determine the optimal solution. In the longest sequence (i.e., sequence 20), only the HE-L-PSO algorithm could determine the optimal structure within 25 predictions. Regarding the overall optimal solutions, the HE-L-PSO algorithm could derive a total of 357 optimal solutions in all experiments, which is higher than those observed for the other algorithms as well as the HEPSO algorithm or the three local search algorithms alone.
Sequences from Table 1.
The three local search algorithms comprise the hill-climbing algorithm, greedy algorithm, and Tabu table.
HE-L-PSO is the combination of HEPSO and three local search algorithms.
Abbreviations (Smith, 2005): COMA, coevolving memetic algorithm; adaptive versions of the COMA were used with the two pivot rules (GComa and SComa), where GComa is the greedy version of the COMA. The versions of the COMA were applied using a randomly created rule in each application (i.e., learning was disabled). SRand, steepest. GRand, greedy. One iteration of SRand or GRand ascent local search was applied. SMA, simple memetic algorithm; SMA using a bit-ipping neighborhood, with one iteration of greedy ascent, was applied. GA, genetic algorithm without local search.
3.4. Mean of optimal fitness
In general, the prediction performance for sequences shorter than 22 amino acids (sequences 1–13) was superior to that for sequences longer than 23 amino acids (sequences 14–20; Table 3). Figure 4 presents statistics for the optimal performance of the algorithms in predicting these longer sequences in the 25 experimental executions. Except for sequence 17, means (standard deviations) for all test sequences were highest (lowest) in the HE-L-PSO algorithm. In addition, for sequences longer than 60, the HE-L-PSO algorithm performed more effectively than did the other tested algorithms such as SGA (Tamjidul Hoque et al., 2006), HGA (Tamjidul Hoque et al., 2006), ERS-GA (Su et al., 2011), and HHGA (Su et al., 2011) (Table 4).
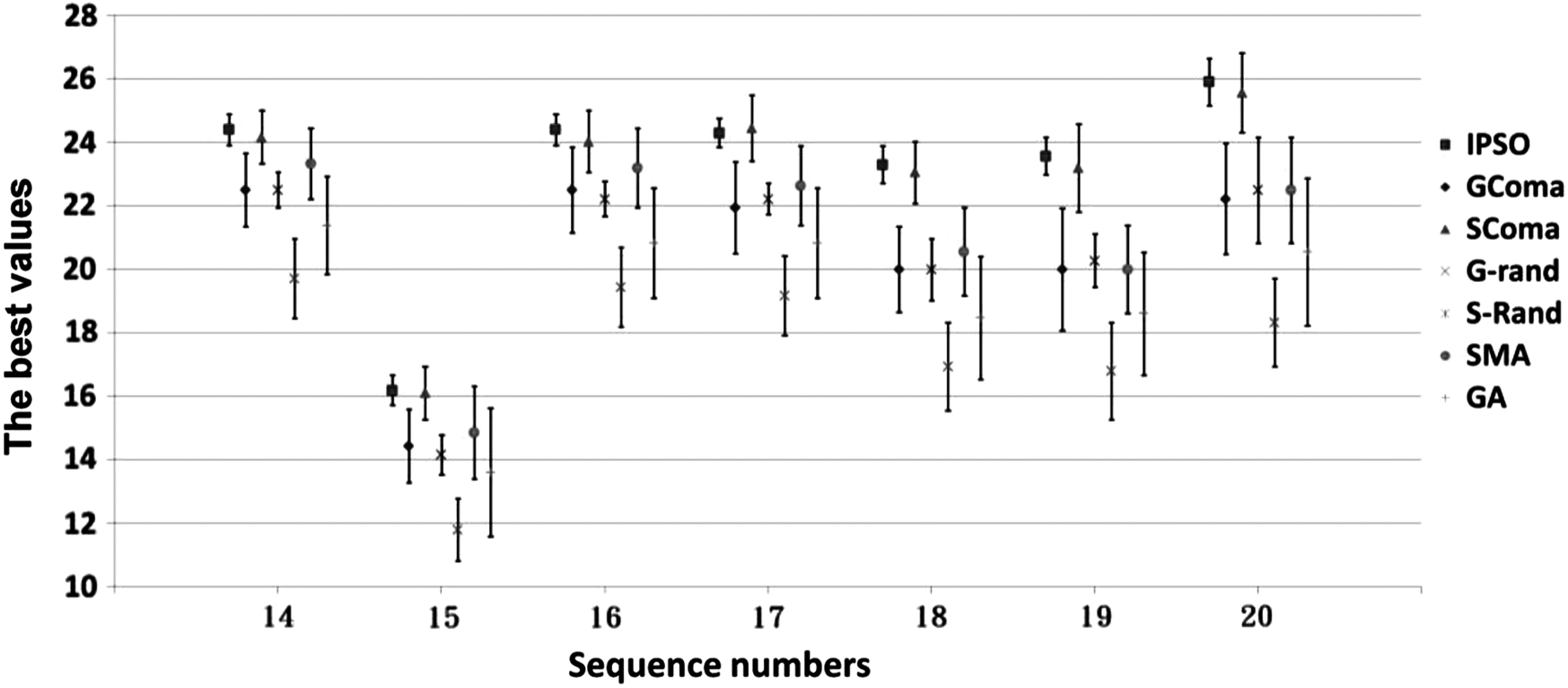
Optimal values analyzed by seven algorithms in the example of sequences 14–20 after 25 experiments. Sequence information is shown in Table 1. Data = mean ± SD (n = 25). SMA, simple memetic algorithm.
H3 is HHH; (PH)2 is PHPH.
P(PH3)2H5P3H10PHP3H12P4H6PH2PHP.
H12(PH)2((P2H2)2P2H)3(PH)2H11
Only the optimal prediction result was reported in the literature without the average value for repetitive experiments.
The values indicate the optimal prediction result and the average value for 25 repetitive experiments.
3.5. Visualization of optimal prediction results
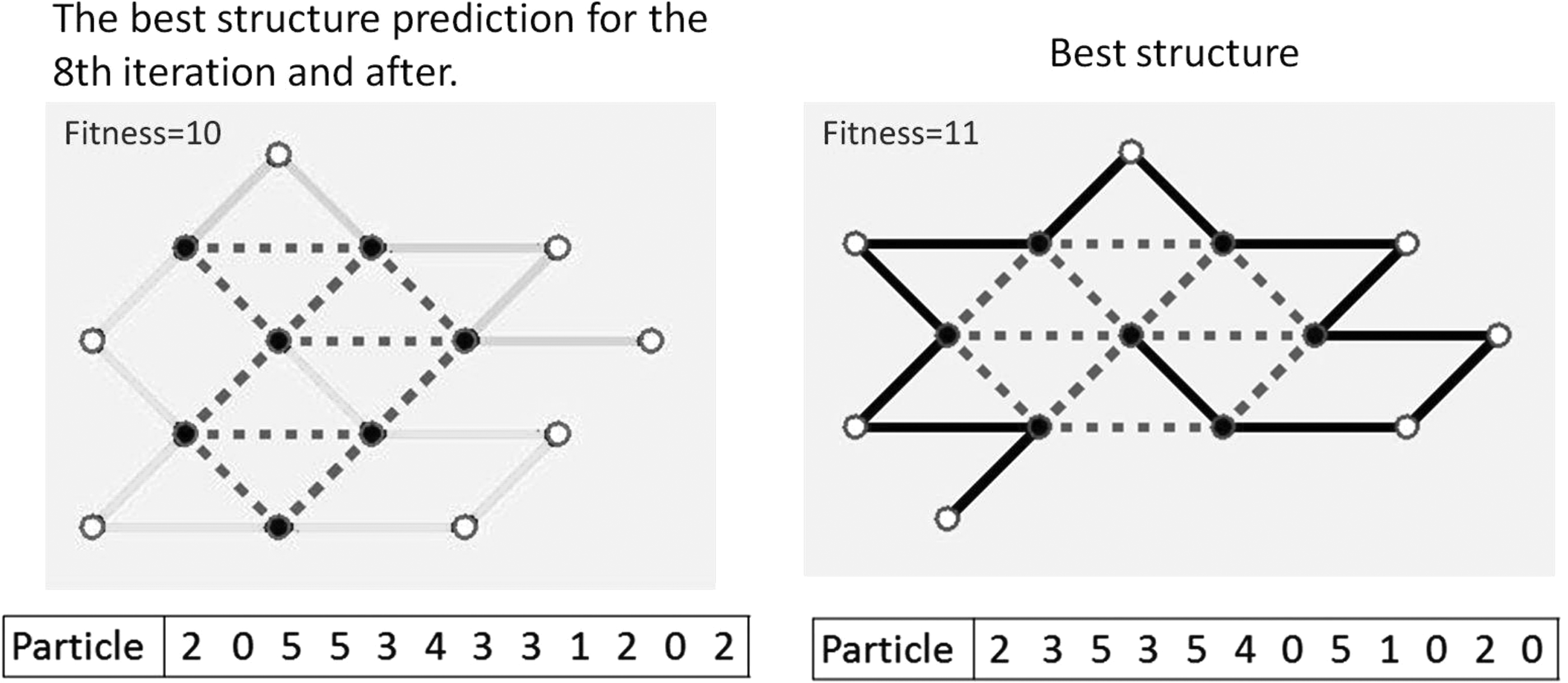
On the basis of the longest sequence (sequence 20 in Table 1), optimal prediction (Table 2) is visualized in Figure 5. The values of hydrophobic interactions in protein folding predicted by the HE-L-PSO algorithm were higher than those of the interactions in protein folding predicted by the HEPSO algorithm or three local search algorithms alone. The optimal HE-L-PSO-based prediction for the 20 amino acid sequences (Table 2) is visualized in Figure 6. The high values of hydrophobic interactions indicate that the predicted structures were closer to the natural folded state. All the HE-L-PSO-predicted protein folding structures possessed a hydrophobic core that is similar to a normal protein folding.
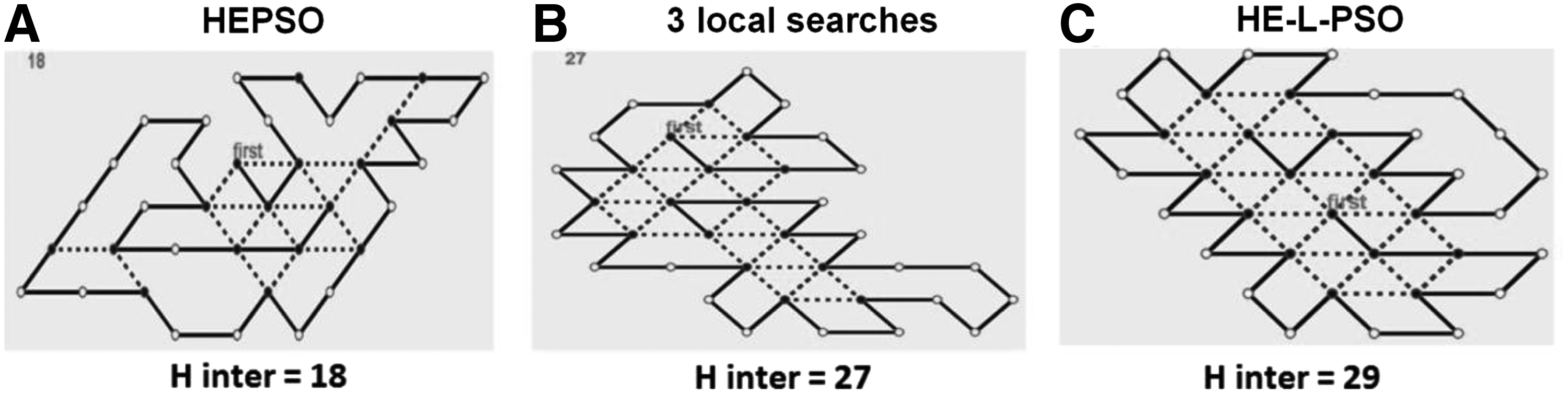
Visualization and comparison of the optimal prediction outcomes for the long sequence predicted using the HEPSO algorithm, three local search algorithms, and HE-L-PSO algorithm. The long sequence is sequence 20 in Table 1 (the longest among all sample sequences). The results are optimal outcomes among 25 tests.

Visualization of the optimal prediction outcome for the 20 amino acid sequences. These results are optimal outcomes of the 25 tests. Twenty sequence samples (Table 1) are marked with S, namely S1–S20. The length (L) of the amino acid sequence ranges from 12 to 37. Black and white points represent hydrophobic and polar amino acids, respectively. All amino acids in the sequence order are connected by solid lines, where the hydrophobic interactions (H inter) are marked with dashed lines. “First” is the starting amino acid for simulation. A higher value of the H inter indicates that the predicted structures are closer to the natural folded state.
4. Discussion
Numerous machine learning algorithms (Gromiha and Huang, 2011), including Monte Carlo algorithms (Mirny and Shakhnovich, 2001; Li et al., 2011), GA (Huang et al., 2005, 2010), ant colony optimization (Shmygelska et al., 2002), the memetic algorithm (Islam and Chetty, 2009), branch and bound (Chen and Huang, 2005; Hsieh and Lai, 2011), the filter-and-fan solution (Rego et al., 2011), and other algorithms listed in Tables 2 and 3, have been proposed for predicting protein folding structures. Optimization algorithms usually converge into an adequate and similar structure; nevertheless, such algorithms are still not the real optimal solution. Moreover, local search algorithms such as the hill-climbing algorithm, greedy algorithm (DeRonne and Karypis, 2006), and Tabu table have been implemented for individually enhancing prediction of protein folding (Lin et al., 2014). The HE-L-PSO algorithm proposed in the current study demonstrates reliable and effective performance in predicting protein folding.
The protein folding problem in the HP model entails determining the optimal structure that maximizes the number of adjacencies between hydrophobic amino acids under the assumption that hydrophobic reactions contribute to embed the free energy for protein folding (Fig. 6). This study applied the HEPSO algorithm to predict protein folding, in addition to using the greedy algorithm, hill-climbing algorithm, and Tabu table in early, medium, and later iterations, respectively, to improve the search ability of the HEPSO algorithm. The hill-climbing algorithm may be insufficient for improving protein folding structures; this is because the particles exhibit poor fitness in the early iteration (Fig. 7, left block). The greedy algorithm is excellent for improving protein folding structures when the protein structures are not dense (Fig. 7, right block). Therefore, when the hill-climbing algorithm does not improve the particle fitness, the greedy algorithm can be applied to improve protein folding structures.
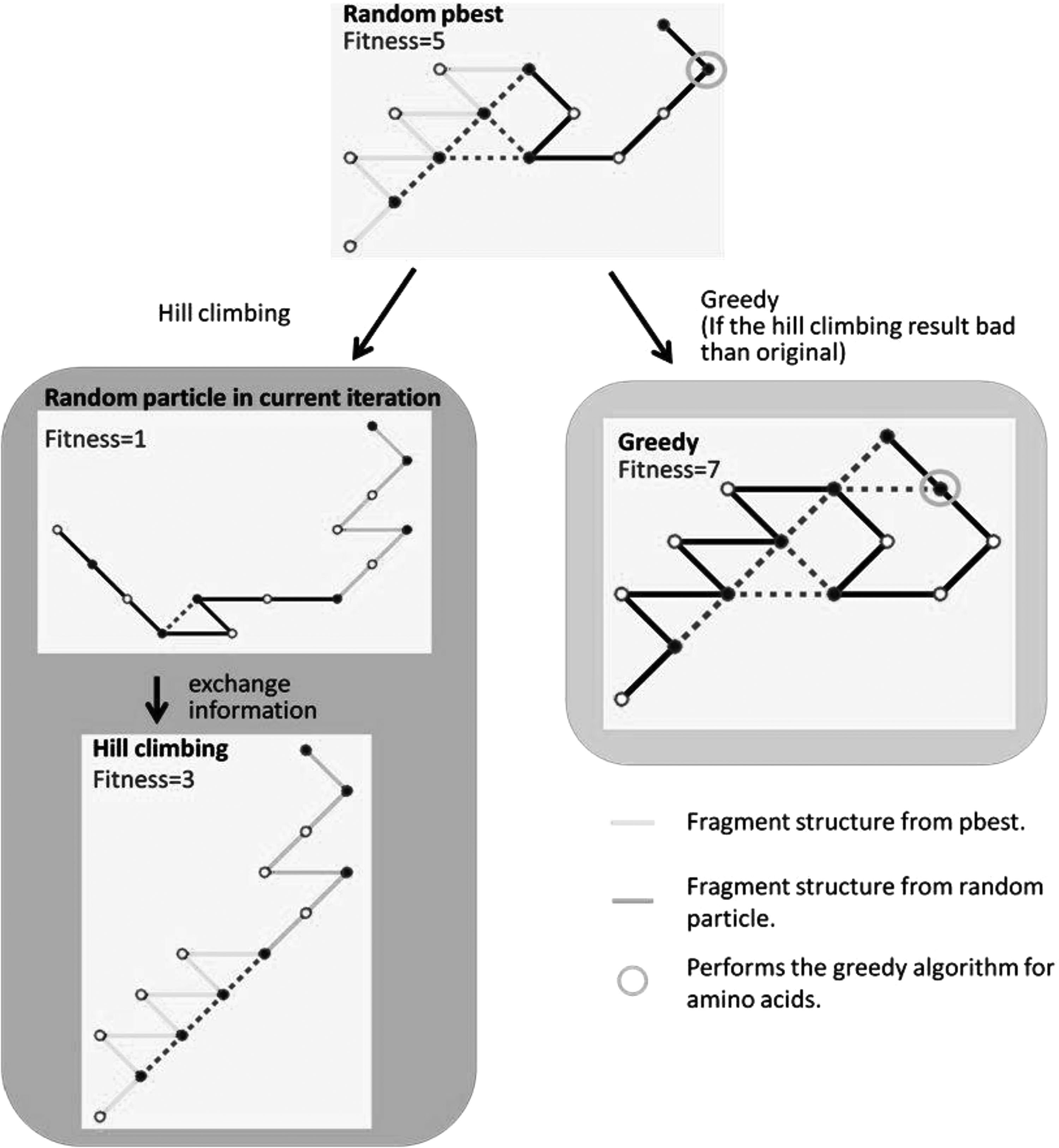
Example of hill climbing performed at a later iteration of the PSO algorithm. Green line represents results derived from a random pbest. Yellow line represents results derived from random particles in the current iteration.
The number of adjacencies between the hydrophobic amino acids of the predicted protein structure for a specific particle can thus be increased along with the number of iterations. In the medium iteration, the hill-climbing algorithm can effectively facilitate particles to detect superior protein structures through an operation that involves exchanging the substructures between two dense protein folding structures (Fig. 8). In the search space, the particles can converge to a local region through numerous generations, a phenomenon that is known as the problem of local optima. The particles face difficulty in escaping from a local optimum because improving the protein folding structures is difficult, particularly because of numerous adjacencies between hydrophobic amino acids (Fig. 9, left). The renewed technique is a powerful evolutionary algorithm that enables particles to escape the local optimum by jumping a longer distance in the search space. However, the particles may still move toward the same search path because of the PSO convergence property. The Tabu table is used to prevent the particles from moving toward the same search path by changing gbest. Therefore, superior results could be obtained for the particles in the new local region (right of Fig. 9).
Example of hill climbing performed at an early iteration of the PSO algorithm and example of the greedy algorithm. Green line represents results derived from a random pbest. Yellow line represents results derived from random particles in the current iteration. Green circle represents the amino acids executing greedy.
A satisfactory structure may not be similar to an optimal structure. The visualization of the structure underneath is the particle content.
Advantages of the proposed HE-L-PSO algorithms are described as follows. (1) High reproducibility: Compared with other algorithms, the HE-L-PSO algorithm was noted to demonstrate a higher number of optimal solutions for 25 executions of the same test (Table 2). (2) High performance in predicting protein folding for long sequences: The HE-L-PSO algorithm is more effective in predicting optimal structures for longer sequences compared with other algorithms (Tables 3 and 4). (3) High stability in predicting protein folding: The HE-L-PSO algorithm was also determined to be stable when predicting optimal protein structures (Fig. 4) because of its short standard deviation for test sequences.
In general, most algorithms for predicting protein folding exhibit lower success rates in long sequences than they do in short sequences. The reason is simple: increasing the sequence length raises the complexity of escaping from the local minima in the protein folding space. Similarly, HE-L-PSO and other algorithms perform comparatively more effectively for short sequences than they do for longer ones. However, the HE-L-PSO algorithm performs more effectively than do the other algorithms tested in Table 3. These results suggest that the HE-L-PSO algorithm is an improved algorithm for predicting protein folding in a triangular lattice model.
In conclusion, this study applied an optimization algorithm (HEPSO) and three local search algorithms to predict protein folding. When used to repeatedly test 20 amino acid sequences 25 times, the proposed HE-L-PSO algorithm demonstrated high performance in predicting both short and long sequences with high reproducibility and stability.
Footnotes
Acknowledgments
This work was supported by funds from the Ministry of Science and Technology, Taiwan (MOST 105-2221-E-151-053-MY2, MOST 103-2221-E-151-029-MY3, MOST 104-2221-E-214-035-MY2, and MOST 104-2320-B-037-013-MY3), and the National Sun Yat-sen University-KMU Joint Research Project (#NSYSU-KMU 105-p022).
Author Disclosure Statement
No competing financial interests exist.