
Abstract
Abstract
Metatranscriptomics studies the transcriptome of all microbial species in a habitat. Removing ribosomal RNA (rRNA) reads in metatranscriptomic data is essential for the study of microbial gene expression. Although several methods are developed, all of them rely on rRNA databases that contain a limited number of known rRNA sequences and cannot work well on rRNA reads from unknown rRNA sequences. To address this problem, we have developed a novel approach called rRNAFilter. Our method can accurately and rapidly remove rRNA reads from metatranscriptomes without any prior knowledge of known rRNA sequences. Compared with two existing approaches, rRNAFilter has shown comparable performance when working on reads from known rRNA sequences and much better performance when dealing with reads from unknown rRNA sequences.
1. Introduction
M
Different experimental and computational approaches have been developed to remove rRNA reads. Several rRNA depletion or mRNA amplification experimental protocols can help to remove rRNA reads (He et al., 2010; Gilbert and Hughes, 2011). However, the metatranscriptomic datasets still contain a large portion of reads from rRNAs after applying these protocols. A few computational methods have also been developed to remove rRNA reads in metatranscriptomes (Kopylova et al., 2012; Schmieder et al., 2012). These methods usually filter rRNA reads by comparing reads with known rRNA sequences in public databases or models derived from known rRNA sequences. Although these methods have shown high accuracy in previous studies (Kopylova et al., 2012; Schmieder et al., 2012), their accuracy depends on the percentage of reads from known rRNA sequences in a metatranscriptomic dataset. Moreover, as shown in this study, these methods cannot work well on rRNA reads from unknown rRNA sequences.
In this study, we developed a novel rRNA filtering tool, rRNAFilter. Our tool does not require any prior knowledge of known rRNA sequences. It filters rRNA reads based on the difference of the frequency of k-mers (k base pairs long DNA segments) in input reads. The rationale is that rRNA reads are much more abundant than non-rRNA reads and k-mers in input reads should be able to distinguish the two types of reads. Compared with two popular methods for removing rRNA reads, rRNAFilter had a much faster speed and at least a comparable accuracy (see Reference 1 for url).
2. Materials and Methods
2.1. Overview of the major steps in rRNAFilter
RRNAFilter applies an expectation-maximization (EM) algorithm (Li and Waterman, 2003; Wang et al., 2015) to k-mers in input reads and separates k-mers of different abundance into different groups (Fig. 1). It then assigns reads comprising k-mers from high-abundant k-mer groups as rRNA reads and reads comprising k-mers from low-abundant k-mer groups as non-rRNA reads. The majority of reads are assigned with high confidence at this step. Next, Markov models are trained with the assigned rRNA reads and non-rRNA reads to represent the common characteristics of the rRNA reads and the non-rRNA reads, respectively. Finally, the unassigned reads are compared with the two trained Markov models to assign the remaining reads.
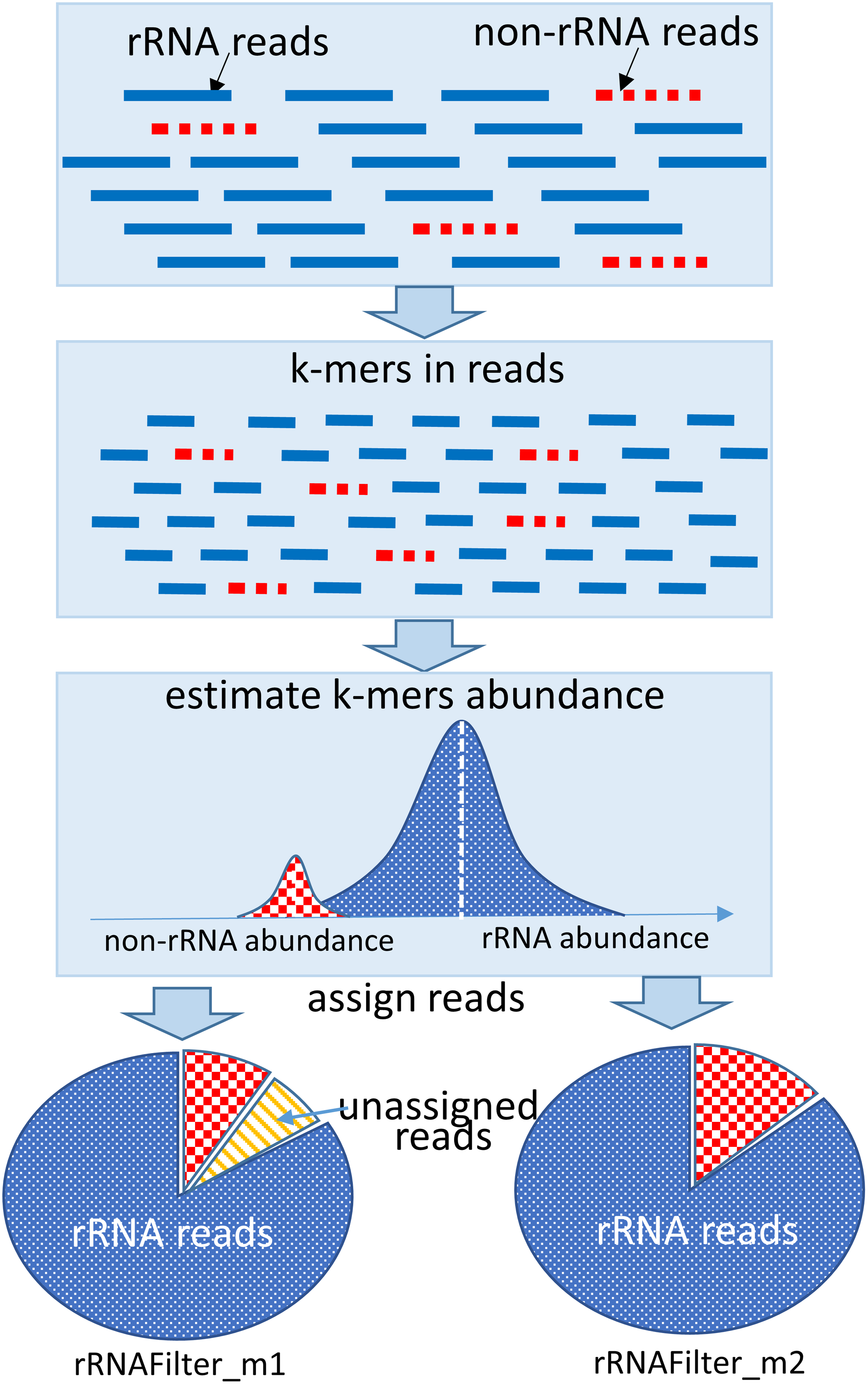
The procedure to assign reads in metatranscriptomic datasets by rRNAFilter. The rRNA reads are represented by solid lines, and the non-rRNA reads are represented by dotted lines. rRNA, ribosomal RNA.
There are two different modes in rRNAFilter. In the first mode, rRNAFilter_m1, it assigns reads by the above procedure without the last two steps. These assigned reads are highly likely rRNA reads or non-rRNA reads. This mode may leave a fraction of reads unassigned. In the second (default) mode, rRNAFilter_m2, rRNAFilter assigns all input reads as either rRNA reads or non-rRNA reads by the above procedure, including the last two steps. The details are in the following.
2.2. EM algorithm
RRNAFilter applies an EM algorithm to separate rRNA reads from non-rRNA reads. The EM algorithm assumes that the frequency of k-mers in input reads,
where
With the above notations, the log complete likelihood function of the observed data X and the missing data
To apply the above EM algorithm, rRNAFilter initializes
The predicted k-mer coverage
2.3. Assign reads with the first mode, rRNAFilter_m1
For each read, rRNAFilter calculates the average frequency of all its k-mers in input reads as the estimated abundance of this read. Since the average abundance of the non-rRNA reads is much smaller than that of rRNA reads, rRNAFilter assumes that k-mers with an abundance close to the smallest estimated abundance,
2.4. Assign remaining reads with the second mode, rRNAFilter_m2
To assign the remaining reads, rRNAFilter evaluates whether an unassigned read is more similar to rRNA reads than to non-rRNA reads. RRNAFilter trains two ninth-order Markov chains using the above grouped rRNA reads and non-rRNA reads to describe their common characteristics, respectively. The ninth-order Markov chain is used based on our previous study (Wang et al., 2016). Note that the Markov chain trained from rRNA reads is more informative than that trained from non-rRNA reads since the number of rRNA reads in the dataset is usually much larger than that of non-rRNA reads. We have also tested lower order Markov chains. The ninth order of Markov chains can in general help to assign rRNA reads more accurately. For each group of reads (rRNA reads or non-rRNA reads), rRNAFilter calculates the stationary and transition probabilities of the ninth-order Markov chain by counting the 9-mer and 10-mer frequencies on both positive and negative strands of the reads assigned to the corresponding read group. A pseudocount, 0.0001, is added to each count to avoid any count to be zero. With the trained Markov chains, the similarity score of a single-end read
2.5. Simulated datasets and real datasets
We evaluated rRNAFilter with nine simulated datasets, each containing 1 million Illumina 100 nucleotide long reads. The first three datasets, one to three, were obtained from a previous study and comprised only rRNA reads (Kopylova et al., 2012). The second three datasets, each of which contained 500,000 rRNA reads and 500,000 mRNA reads, had reads randomly selected from the reads used in the same previous study (Kopylova et al., 2012). The last three datasets contained only rRNA reads generated from rRNA sequences randomly selected from a more comprehensive rRNA database. To make this a more comprehensive database, we combined nonredundant rRNA sequences from the following databases: SILVA (Quast et al., 2013), HOMD (Chen et al., 2010), Greengenes (DeSantis et al., 2006), and RDP (Cole et al., 2009). A fraction of rRNA sequences in these more comprehensive databases was not included in the rRNA database used by SortMeRNA (Kopylova et al., 2012). The rRNA reads were then simulated by the MetaSim tool with the randomly selected rRNA sequences (Richter et al., 2008).
We also tested rRNAFilter with six publicly available metatranscriptomic datasets. Two datasets (SRR106861 and SRR013513) were from the study of SortMeRNA (Kopylova et al., 2012) and four additional datasets (SRR1013758, SRR933607, SRR3441864, SRR3441820) were downloaded from www.ncbi.nlm.nih.gov/sra and preprocessed by the tool PRINSEQ (Schmieder and Edwards, 2011), which can filter, reformat, or trim reads, such as removing sequence copies, sequences comprising N's, and low-quality sequences.
2.6. Comparison with other tools
We compared rRNAFilter with two popular tools, SortMeRNA and riboPicker (Kopylova et al., 2012; Schmieder et al., 2012). Both tools have shown to have high recall. They were run with the default setting by using the same rRNA reference database from SortMeRNA.
3. Results
3.1. RRNAFilter reliably identified rRNA reads on simulated datasets
To see how well rRNAFilter could identify rRNA reads, we tested it on the first three simulated datasets. These datasets were from the study of SortMeRNA, with all reads in each dataset from rRNA sequences. We calculated the recall to measure the accuracy of rRNAFilter as the precision would be one on these datasets. To compare, we also applied two popular tools, SortMeRNA and riboPicker (Kopylova et al., 2012; Schmieder et al., 2012), to the same datasets. The recall of the three methods on the three datasets was shown in Table 1. Note that since rRNAFilter_m1 did not consider all input reads, we only used all its assigned reads to calculate its recall. On average, rRNAFilter_m2 had a 3.14% higher recall than riboPicker and a 0.22% lower recall than SortMeRNA. Moreover, rRNAFilter_m1 confidently filtered more than 74% of total rRNA reads in each dataset. Its recall was 0.1% higher than that of SortMeRNA and 3.47% higher than that of riboPicker. This indicated that most reads in a metatranscriptomic dataset could be reliably filtered by rRNAFilter with high confidence and without any reference database.
The recalls are provided for the first and last groups of three datasets since the precision is one for all datasets for all tools. The recall/precision are provided for the second group of three datasets in the format of recall/precision.
rRNA, ribosomal RNA.
We further tested rRNAFilter with the second three datasets. Although the reads in these three datasets were also from the study of SortMeRNA, half of the reads in each dataset were from non-rRNA sequences. We calculated both recall and precision for the three methods on these three datasets (Table 1). We found that rRNAFilter_m2 had the same recall as SortMeRNA, which was higher than the recall of riboPicker. Its precision was better than that of riboPciker while slightly lower than SortMeRNA. Moreover, rRNAFilter_m1 had the same recall as SortMeRNA, but a better precision than SortMeRNA and riboPicker.
Since rRNAFilter does not depend on known rRNA sequences, while other methods do, we further compared the three methods on three additional datasets that contained rRNA reads from rRNA sequences that may not be used to train SortMeRNA and riboPicker. We found that SortMeRNA and ribopicker could hardly work on these datasets, while rRNAFilter had similar performance as that on the above six datasets. This demonstrated that rRNAFilter can filter rRNA reads without relying on any rRNA reference database. It also suggested the wide applications of rRNAFilter to the current metatranscriptomic datasets, which commonly contain unknown rRNA sequences.
3.2. RRNAFilter performs well on experimental datasets
We also tested the tools on six publicly available metatranscriptomic datasets, including two (SRR106861 and SRR013513) from the study of SortMeRNA (Materials and Methods section). With not much annotation of the reads in these datasets, we evaluated the tools using BLASTN (Camacho et al., 2009) with the default parameters and the same rRNA reference database used by SortMeRNA. The rRNA reads identified by BLASTN were considered as true rRNA reads.
RRNAFilter showed a similar recall as SortMeRNA and a better recall than riboPicker (Table 2). On average, rRNAFilter_m2 had a 3.18% higher recall than riboPicker and a 0.007% lower recall than SortMeRNA. RRNAFilter_m1 confidently filtered more than 72% of reads, with its average recall 3.34% higher than that of riboPicker and 0.04% higher than that of SortMeRNA. This indicated that rRNAFilter_m1 could be used to initially filter most rRNA reads in a metatranscriptomic dataset with high accuracy and fast speed. For these datasets, rRNAFilter_m1 showed a higher precision than SortMeRNA for three of the six datasets, while rRNAFilter_m2 showed a lower precision than the other two methods for almost all datasets.
For each tool, the number of identified rRNA reads is provided, followed by the recall/precision of the tool.
The lower precision of rRNAFilter_m2 may be due to the fact that there exist unknown rRNA reads that could not be detected by the above procedure. To see whether this hypothesis was true, we compared the predicted rRNA reads with rRNA sequences in a more comprehensive database (Materials and Methods section). We mapped these predicted rRNA reads by each method to this new rRNA database by BLASTN with a smaller word_size parameter, 7. We found that a large number of predicted rRNA reads that were not identified as rRNA reads by the default BLASTN were detected as rRNA reads by the new procedure (Table 3).
The #additionally predicted rRNA reads are the number of predicted rRNA reads that cannot be mapped to rRNA sequences with the default BLASTN and the default rRNA database. The #new mapped rRNA reads are the number of additionally predicted rRNA reads that are mapped to rRNA sequence with the new BLASTN parameter and the more comprehensive rRNA database. The percentages shown in the fourth and fifth column are the percentages of reads from SortMeRNA that are shared by the reads from rRNAFilter.
To test the significance of the observed large number of additional rRNA reads in each dataset, we also mapped the predicted non-rRNA reads in each dataset with the same new procedure. By Fisher's exact test, we found that the observed number of predicted rRNA reads that were mapped to rRNA sequences in this new database is not by chance (p < 2.9e-122), suggesting that these additionally mapped rRNA reads were likely true rRNA reads.
Since the above new procedure made sense in mapping unknown rRNA reads, we further compared how well rRNAFilter_m2 and SortMeRNA predicted the additional rRNA reads (Table 3). We did not consider riboPicker since SortMeRNA was superior to riboPicker on all datasets. In all six datasets, much more number of predicted rRNA reads by rRNAFilter could be mapped to rRNA sequences in the more comprehensive rRNA database than that by SortMeRNA. Moreover, almost all predicted rRNA reads that could be mapped to rRNA sequences in the new database by SortMeRNA were also predicted by rRNAFilter. Interestingly, in all six datasets, the percentage of the SortMeRNA-predicted rRNA reads that were also predicted by rRNAFilter was smaller than the percentage of the SortMeRNA-predicted rRNA reads that could be mapped to new rRNA sequences and were also predicted by rRNAFilter, supporting the good quality of the predicted rRNA reads by rRNAFilter. It is also worth pointing out that rRNAFilter_m2 had a higher recall than SortMeRNA, with the additionally mapped reads (Supplementary Table S1).
We also compared the speed of the tools. Because SortMeRNA and riboPicker compare reads with known rRNA databases, they are time-consuming. On the other hand, rRNAFilter, which does not compare sequences, has a much faster speed, about 26 times faster than BLASTN, 5 times faster than SortMeRNA, and 6 times faster than riboPicker (Fig. 2).
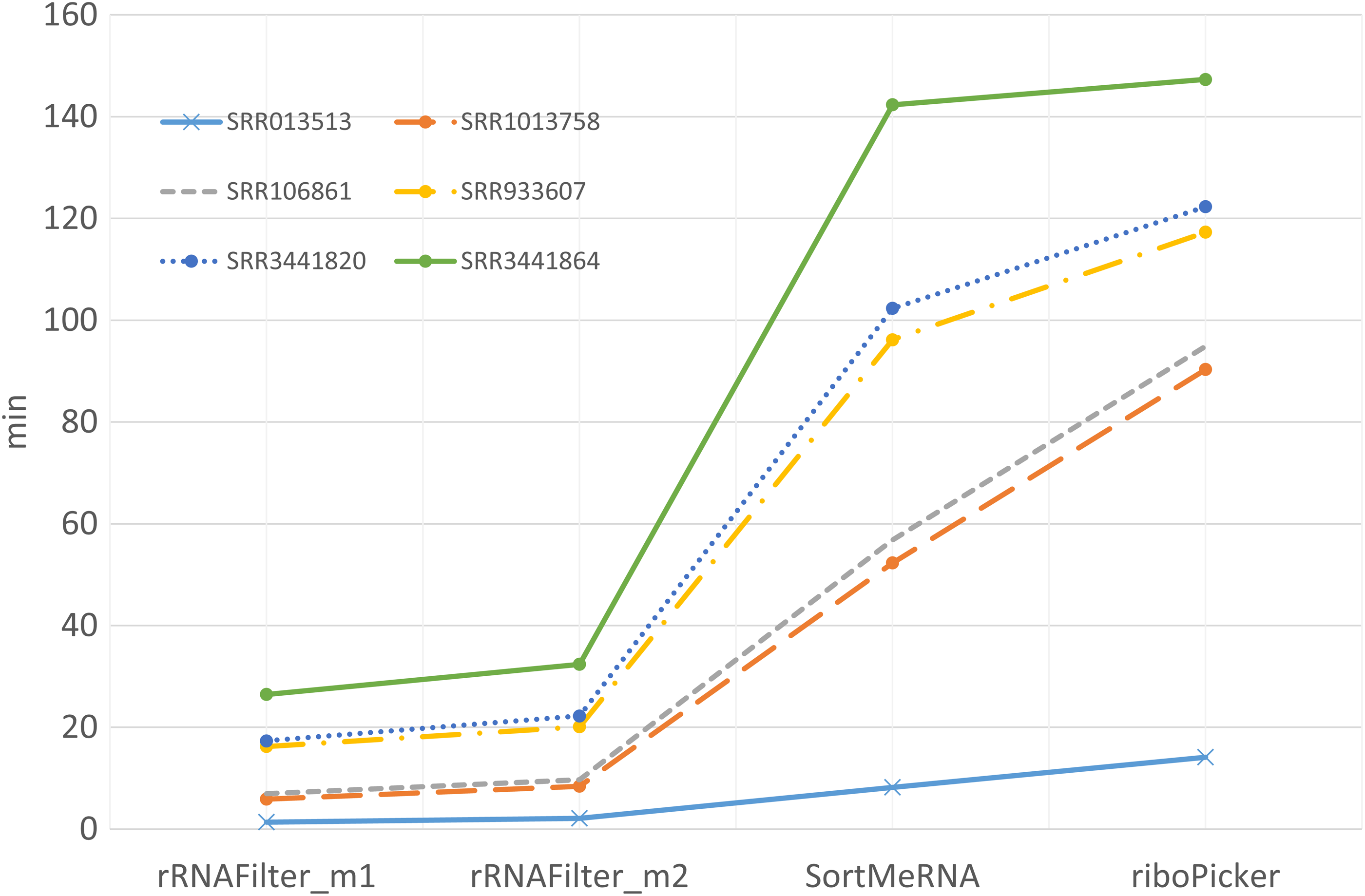
The running time comparison on six experimental datasets.
4. Discussion and Conclusion
We developed a novel approach rRNAFilter to remove rRNA reads from metatranscriptomic datasets. Our method is different from existing approaches that rely on rRNA reference databases. It considers the abundance difference of rRNA reads and non-rRNA reads to separate them, without depending on any reference database. Compared with other methods, rRNAFilter has a comparable accuracy and much faster speed.
Due to the limitation of the current rRNA databases, rRNAFilter showed a lower precision than other methods. This was especially evident in the experimental dataset, SRR1013758. Compared with a more comprehensive rRNA database, we demonstrated that rRNAFilter indeed predicted a much larger number of true rRNA reads than existing methods and its precision was significantly improved with the more comprehensive database (Supplementary Table S1).
We also want to point out that although the additionally predicted rRNA reads may be from real rRNA sequences, some of them may be from unknown abundant mRNA, or other types of RNA sequences. In fact, we noticed that some predicted rRNA reads by rRNAFilter and SortMeRNA can be mapped to the ribosomal protein gene (RPG) mRNA sequences (Nakao et al., 2004) in the six experimental datasets (Supplementary Table S2). We also observed that slightly fewer rRNA reads predicted by rRNAFilters were similar to RPG mRNA sequences than those predicted by SortMeRNA, although rRNAFilter predicted much more rRNA reads than SortMeRNA.
In practice, one can run both rRNAFilter and existing tools and combine the predicted rRNA reads. When run existing tools, we recommend to run rRNAFilter with the first mode first and then apply the existing tools to the remaining unassigned reads. In this way, we may be able to remove rRNA reads from unknown rRNA sequences confidently. At the same time, we will be able to employ the known rRNA sequence information to further remove rRNA reads.
Footnotes
Acknowledgments
This work has been supported by the National Science Foundation [Grant Nos. 1356524, 1149955, and 1218275] and the National Institutes of Health [Grant No. 2R01HL048044]. Funding for open access charge was provided by The National Science Foundation grant 1218275.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.