
Abstract
Abstract
With the rapid growth of known protein 3D structures in number, how to efficiently compare protein structures becomes an essential and challenging problem in computational structural biology. At present, many protein structure alignment methods have been developed. Among all these methods, flexible structure alignment methods are shown to be superior to rigid structure alignment methods in identifying structure similarities between proteins, which have gone through conformational changes. It is also found that the methods based on aligned fragment pairs (AFPs) have a special advantage over other approaches in balancing global structure similarities and local structure similarities. Accordingly, we propose a new flexible protein structure alignment method based on variable-length AFPs. Compared with other methods, the proposed method possesses three main advantages. First, it is based on variable-length AFPs. The length of each AFP is separately determined to maximally represent a local similar structure fragment, which reduces the number of AFPs. Second, it uses local coordinate systems, which simplify the computation at each step of the expansion of AFPs during the AFP identification. Third, it decreases the number of twists by rewarding the situation where nonconsecutive AFPs share the same transformation in the alignment, which is realized by dynamic programming with an improved transition function. The experimental data show that compared with FlexProt, FATCAT, and FlexSnap, the proposed method can achieve comparable results by introducing fewer twists. Meanwhile, it can generate results similar to those of the FATCAT method in much less running time due to the reduced number of AFPs.
1. Introduction
T
In the past three decades, many protein structure alignment methods have been proposed (Altschul et al., 1990; Ortiz et al., 2002; Krissinel and Henrick, 2004; Razmara et al., 2012). Based on different ways in finding optimal alignment, protein structure alignment methods can be divided into three groups: aligned fragment pair (AFP)-based methods, distance matrix-based methods, and other methods. The AFP-based methods first divide protein structures into different fragments and then identify the similar structure fragment pairs coming from two different protein structures, which form the set of AFPs. Then, the AFPs are chained with defined constraints to produce the optimal alignment result. For example, CE (Shindyalov and Bourne, 1998) and FATCAT (Ye and Godzik, 2003) are both AFP-based methods. The distance matrix-based methods first calculate distance matrices of substructures according to 3D coordinates of each protein, and then match similar substructures according to their distance matrices. A typical distance matrix-based method is DALI (Holm and Sander, 1993). The third group of structure alignment methods uses different approaches from the former two. For example, DeepAlign (Wang et al., 2013) aligns two protein structures using not only spatial proximity of equivalent residues but also evolutionary relationship and hydrogen-bonding similarities. The LGA (Zemla, 2003) method detects the regions of local and global structure similarities between proteins according to LCS (longest continuous segments) and GDT (global distance test).
Generally, the AFP-based structure alignment methods have a special advantage over the other methods: the AFP-based methods can balance between global structure similarities and local structure similarities, while other methods mainly focus on the global structure similarities during the alignment. According to whether twists are introduced, protein structure alignment can be divided into two categories: rigid structure alignment and flexible structure alignment. CE is a typical example of rigid structure alignment method, which treats protein structures as rigid bodies (Shindyalov and Bourne, 1998). It aligns protein structures by chaining the consecutive AFPs without twists. Considering recent evidence supporting the idea that some proteins exist in different forms in different environments (Holmes, 2009; Godshall et al., 2013; Wang et al., 2014), rigid structure alignment methods may fail to identify the structure similarities that have gone through conformational changes, while flexible structure alignment methods can remove the above limitation by introducing twists in the structure alignment. Until now, many flexible structure alignment methods, such as FlexProt (Shatsky et al., 2002, 2004), FATCAT, and FlexSnap (Salem et al., 2010), have been developed.
In this article, we propose a method for protein structure alignment based on variable-length AFPs. The method is called Flexible protein structure alignment using Variable-length Aligned Fragment Pairs (FlexVAFP). Different from other flexible structure alignment methods such as FATCAT, the proposed method owns three main advantages: first, it can automatically adjust the sizes of AFPs according to local structure similarities, which can not only produce improved representation of the local structure similarities but also can reduce the total number of AFPs. Second, the proposed method uses local coordinate systems in identifying and expanding AFPs, which simplify the computation at each step of the expansion of AFPs during the AFP identification. Third, the proposed FlexVAFP method can decrease the number of twists by rewarding the situation where nonconsecutive AFPs share the same transformation in the alignment, which is realized by dynamic programming with an improved transition function.
The initial results of the proposed method are already reported in a conference article (Hu and Yonggang, 2015). In this study, the method is further evaluated with more structure alignment results; the efficiency of the method is shown by deriving a relationship between the running time and the number of AFPs; and more discussions about the method and its main features are included.
The rest of this article is organized as follows. First, we will describe how to implement the method in detail in Section 2. Second, we will assess and discuss the performance of the proposed method by comparing our method against other structure alignment methods in Section 3. Finally, we will conclude the article in Section 4.
2. Methods
In this section, we will explain our method clearly and state how to implement it in detail.
2.1. Definition of AFP
In the algorithm proposed in this article, AFP represents a local match of two protein fragments from two different proteins under consideration. Given two proteins called protein A and protein B, the AFPk is an aligned match of two consecutive protein fragments coming from protein A and protein B. The starting positions of AFPk in the two proteins are bA(k) and bB(k) and ending positions are eA(k) and eB(k).
To identify high-quality AFPs, length constraint and root mean square deviation (RMSD) constraint of AFPs are defined. First, it requires that the length of AFP is not less than the minimum length Lmin that is set empirically. Second, the maximum value of an AFP RMSD is also restricted to a threshold parameter, THRMSD.
The quality of AFP is measured by the score derived from the following equation:
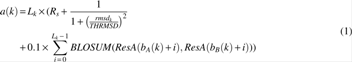
where Lk is the length of AFPk; Rs is a coefficient; rmsdk is the RMSD of AFPk; THRMSD is the RMSD threshold; and ResA(bA(k) + i) and ResB(bB(k) + i) are residues of the (bA(k) + i)th and the (bB(k) + i)th in protein A and protein B, respectively; BLOSUM is the widely used amino acid substitution matrix, BLOSUM62 (Henikoff and Henikoff, 1992).
2.2. Process of producing AFPs
Before producing AFPs, the local coordinates of Cα atoms of residues starting from the 4th residue in protein A and protein B are generated (Fig. 1). According to the locations of Cαi−3, Cαi−2, and Cαi−1 atoms, the local coordinate system centered at Cαi−1 is established. Then, the local coordinates
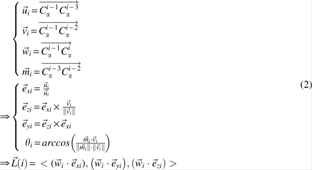
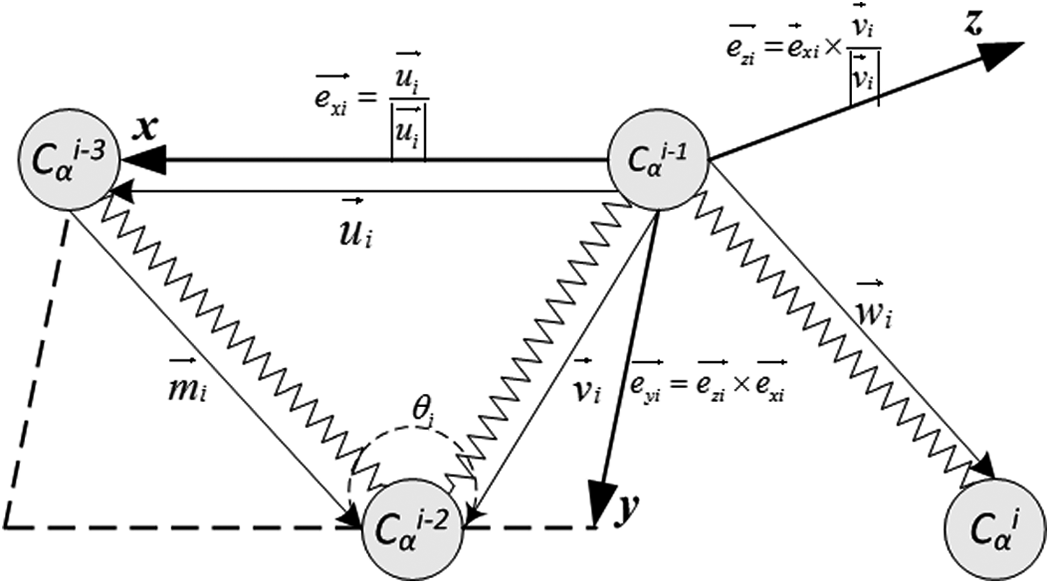
The generation of a local coordinate system for C α i atom.
The process for producing an AFP in the proposed method, FlexVAFP, is shown in Figure 2. It can be divided into four steps: first, it computes the local coordinates of every Cα atom (Equation 2). Second, it looks for the starting residue pair (called the seed) of a possible AFP. To avoid the overlap between AFPs, the starting residue pair cannot exist in other AFPs. Then, conditions,
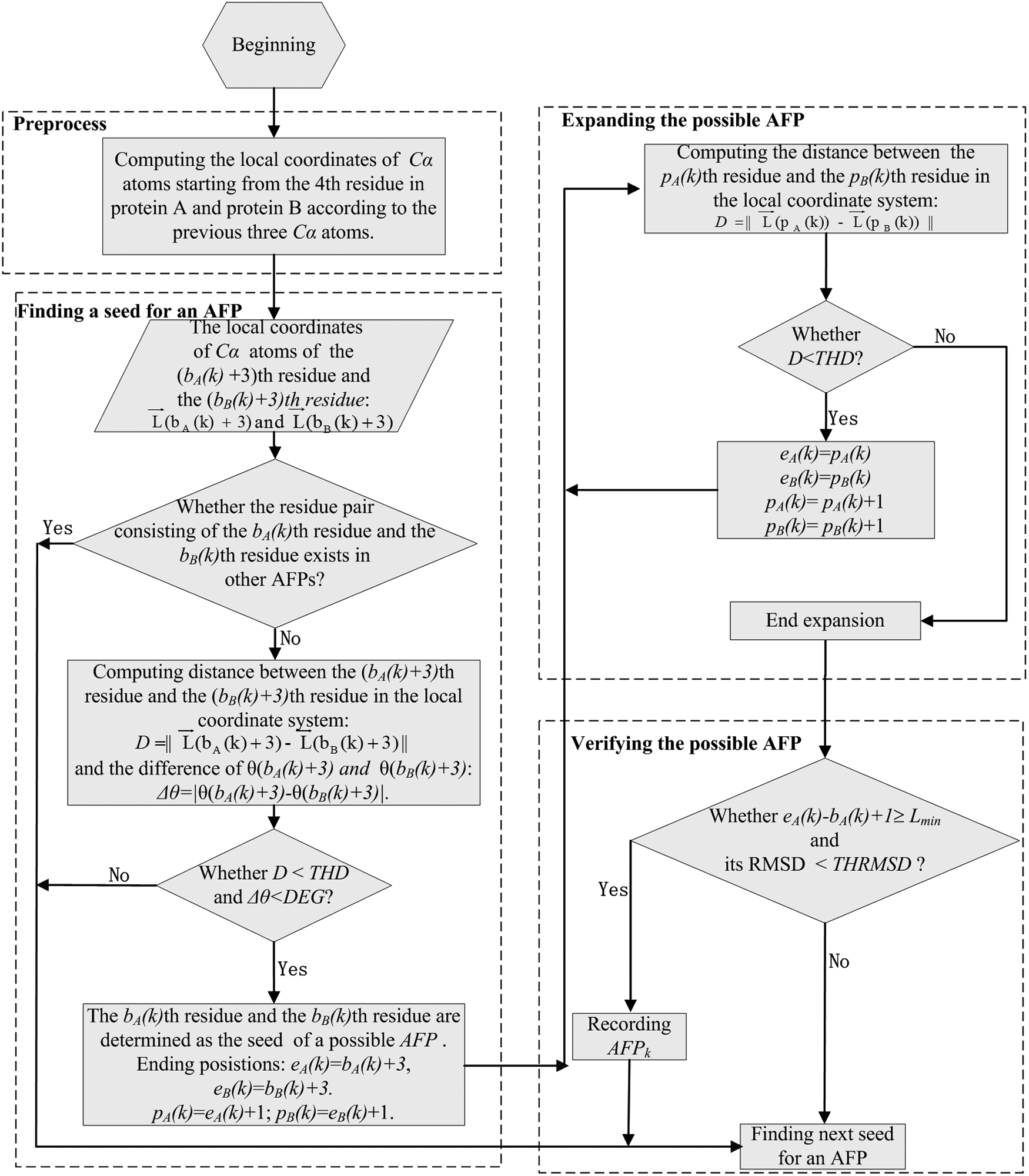
The process of identifying an AFP in the FlexVAFP method. AFP, aligned fragment pair.
2.3. Connecting AFPs by dynamic programming with an improved transition function
To avoid overlap, AFPm and AFPn can be connected if the following conditions are satisfied:
where eA(m) and eB(m) represent the ending positions of AFPm in protein A and protein B, respectively; bA(n) and bB(n) represent the starting positions of AFPn, respectively.
In the proposed method, FlexVAFP, AFPs meeting the condition (Equation 3) are chained by dynamic programming. The connection score from AFPm to AFPn is given by
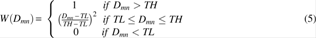
where Pt is the maximum penalty for the connection; Dmn is the RMSD for aligning both AFPm and AFPn; W(Dmn) is a measure to evaluate the quality of the connection from AFPm to AFPn; F(p,q) does the same work by examining the gaps between the two AFPs; TH is a threshold to determine whether AFPm and AFPn can be linked by introducing a twist; TL is a threshold for penalizing a connection from AFPm to AFPn; p is the number of mismatched residues; q is the number of gaps; Ms is the penalty involved with mismatched residues; and Mg is used to penalize gaps.
To produce the optimal alignment result, a dynamic programming algorithm is used. To reward the situation where nonconsecutive AFPs share the same transformation in the alignment, the transition function of dynamic programming is improved. The transition function S(n) denotes the best score ending at AFPn:


where a(n) is the score of AFPn (Equation 1); OPTm represents the best partial alignment path formed by the AFPs ending at AFPm, which has the highest score; and P(m→n) represents the best connection score found from an AFP in the OPTm to AFPn.
The number of twists required to form OPTn is
where T(n) and T(m) are number of twists introduced to form OPTn and OPTm, respectively; t(m→n) is 1 if a new twist is introduced to produce OPTn by adding AFPn to OPTm, otherwise t(m→n) is 0.
By using the improved transition function defined in Equations 7 and 8, the FlexVAFP method can reduce the number of twists by rewarding nonconsecutive AFPs, which share the same transformation in the alignment. For example, if the best partial alignment ending at AFPm is OPTm = {…, AFPi, …, AFPm}, AFPn is the next candidate AFP for concatenation, which satisfies Equation (3), Dmn > TH, and Din ≤ TH; while a twist is needed to connect AFPn with AFPm in a traditional AFP-based method such as FATCAT, no twist is needed to connect AFPn with AFPm in our method because AFPi and AFPn share the same transformation.
2.4. Postprocessing
The postprocessing is executed after achieving the optimal correspondence of Cα atoms between the two proteins by dynamic programming. The FlexVAFP method will produce different number of transformation matrices according to the different number of twists introduced. To produce high-quality global alignment, the RMSD of aligned fragment pairs sharing the same transformation matrix is required to be less than RMSDmax. Suppose that its RMSD is larger than RMSDmax, the FlexVAFP method will introduce a twist until the condition is satisfied. The iterative refinement process is similar to that used by ProSup (Lackner et al., 2000).
3. Results
To intensively evaluate the proposed method, FlexVAFP, it is compared with both rigid structure alignment methods and flexible structure alignment methods. A rigid version of FlexVAFP is also produced to objectively and fairly compare FlexVAFP with rigid structure alignment methods. The rigid version of FlexVAFP (FlexVAFPr) performs essentially the same as FlexVAFP, except that no twist is introduced during the process of chaining AFPs. All the parameters used in our experiments are listed in Table 1.
3.1. Comparison with other rigid structure alignment methods
FlexVAFPr is compared with rigid structure alignment methods, DALI, CE, and ProSup, on several pairs of protein structures described as difficult in the literature (Ye and Godzik, 2003). FlexVAFPr performs well in comparing distantly similar protein structures compared with the performance of CE, DALI, and ProSup (Table 2). For instance, in the comparison of proteins, 1BGE:B and 2GMF:A, FlexVAFPr obtains 95 aligned residues with a lower RMSD of 3.10, while CE gets 94 aligned residues with an RMSD of 4.1, DALI produces 98 aligned residues with an RMSD of 3.5, and ProSup obtains 87 aligned residues with an RMSD of 2.4. In a few particular cases, FlexVAFPr can achieve better results than the three different methods. For example, in the comparison of proteins, 1FXI:A and 1UBQ:_, FlexVAFPr finds 61 aligned residues with an RMSD of 2.85; however, ProSup gets 54 aligned residues with an RMSD of 2.6. Therefore, these results also reflect that the way of identifying AFPs in FlexVAFP is reasonable and effective.
The data for CE, DALI, and ProSup are from literature (Lackner et al., 2000).
S represents the size of aligned residues.
R represents the RMSD of all aligned residues in the unit of Å.
3.2. Comparison with other flexible structure alignment methods
To demonstrate that the FlexVAFP method usually introduces fewer twists, it is compared with three different flexible structure alignment methods, FlexProt, FATCAT, and FlexSnap, on FlexProt dataset (Shatsky et al., 2004). The results are shown in Table 3.
The data for FlexProt, FATCAT, and FlexSnap are from literature (Salem et al., 2010).
S represents the size of the aligned residues.
R represents the RMSD of all aligned residues.
T is the number of twists.
According to the experimental results in Table 3, the proposed method, FlexVAFP, achieves competitive results against the three other methods judged from the number of aligned residues and the RMSD. For instance, in the comparison of proteins, 1MCP:L and 4FAB:L, a hinge is detected by FlexVAFP, which results in a structure alignment of 216 aligned positions with an RMSD of 1.2, while the results of the three other methods are 218 aligned residues with an RMSD of 1.93 (FlexProt), 217 aligned residues with an RMSD of 1.40 (FATCAT), and 217 aligned residues with an RMSD of 1.49 (FlexSnap) with the same number of twists. In all alignment results about the sixteen pairs of proteins in Table 3, the total numbers of twists introduced in the four methods are 30 (FlexProt), 20 (FATCAT), 31 (FlexSnap), and 16 (FlexVAFP), respectively. It can be seen from Table 3 that the number of twists introduced by FlexVAFP is not more than that of the other three different methods in all sixteen alignment results of Table 3, except the result for proteins, 1B9W:A and 1DAN:L.
To further illustrate that the FlexVAFP method can produce comparable alignment results with fewer twists, 25 pairs of protein structures are selected from the database of the literature as a test dataset (Gerstein and Krebs, 1998). The performance of FlexVAFP on the dataset is compared with the three other methods and the results are listed in Table 4.
The data for FlexProt and FATCAT are from their online servers; the data for FlexSnap are from its stand-alone version.
The results in Table 4 also demonstrate that the method, FlexVAFP, can achieve comparable results compared with the other three different methods, FlexProt, FATCAT, and FlexSnap, by introducing fewer twists. In all 25 alignment results, FlexProt finds 7947 aligned residues in total with the sum of RMSD 62.03; FATCAT obtains 8220 aligned residues in total with the sum of RMSD 59.16; FlexSnap gets 8084 aligned residues in total with the sum of RMSD 44.22; and FlexVAFP matches 8076 aligned residues in total with the sum of RMSD 46.4. Compared with FlexProt and FATCAT, both FlexSnap and FlexVAFP achieve comparable aligned residues with lower RMSD. The total numbers of twists introduced by the four methods are 42 (FlexProt), 14 (FATCAT), 36 (FlexSnap), and 17 (FlexVAFP), respectively. Accordingly, compared with FlexSnap, FlexVAFP produces alignment with comparable length and RMSD while introducing fewer twists.
It is clearly seen from Tables 3 and 4 that total number of twists introduced in FlexVAFP is lower than that of FlexProt and FlexSnap. The reason why FlexVAFP can obtain competitive results by introducing lower number of twists is that FlexVAFP allows nonconsecutive AFPs to share the same transformation during the concatenation (Fig. 3).

The alignment results between proteins, 1AKE:A and 2AK3:A, generated by
From Figure 3, it is clear that both FlexProt and FATCAT introduce two twists in the alignment of proteins, 1AKE:A and 2AK3:A, while FlexVAFP only introduces one twist in the alignment of the same protein pair. It can be seen that the sections separated by the green section and the purple section in Figure 3a and b are considered as a single alignment section, which shares the same transformation by FlexVAFP.
3.3. Comparison of FlexVAFP and FATCAT about the number of AFPs
To show that the running time of the AFP-based structure alignment method strongly depends on the number of AFPs, the AFP identification process in FlexVAFP is modified to produce nine different variations of the method based on constant-length AFPs (with the length of AFP required to be 6, 7, 8, 9, 10, 11, 12, 13, and 14, respectively). Then, these nine methods are compared with the original FlexVAFP method. The relationship between the running time and the number of AFPs generated by the methods is shown in Figure 4.
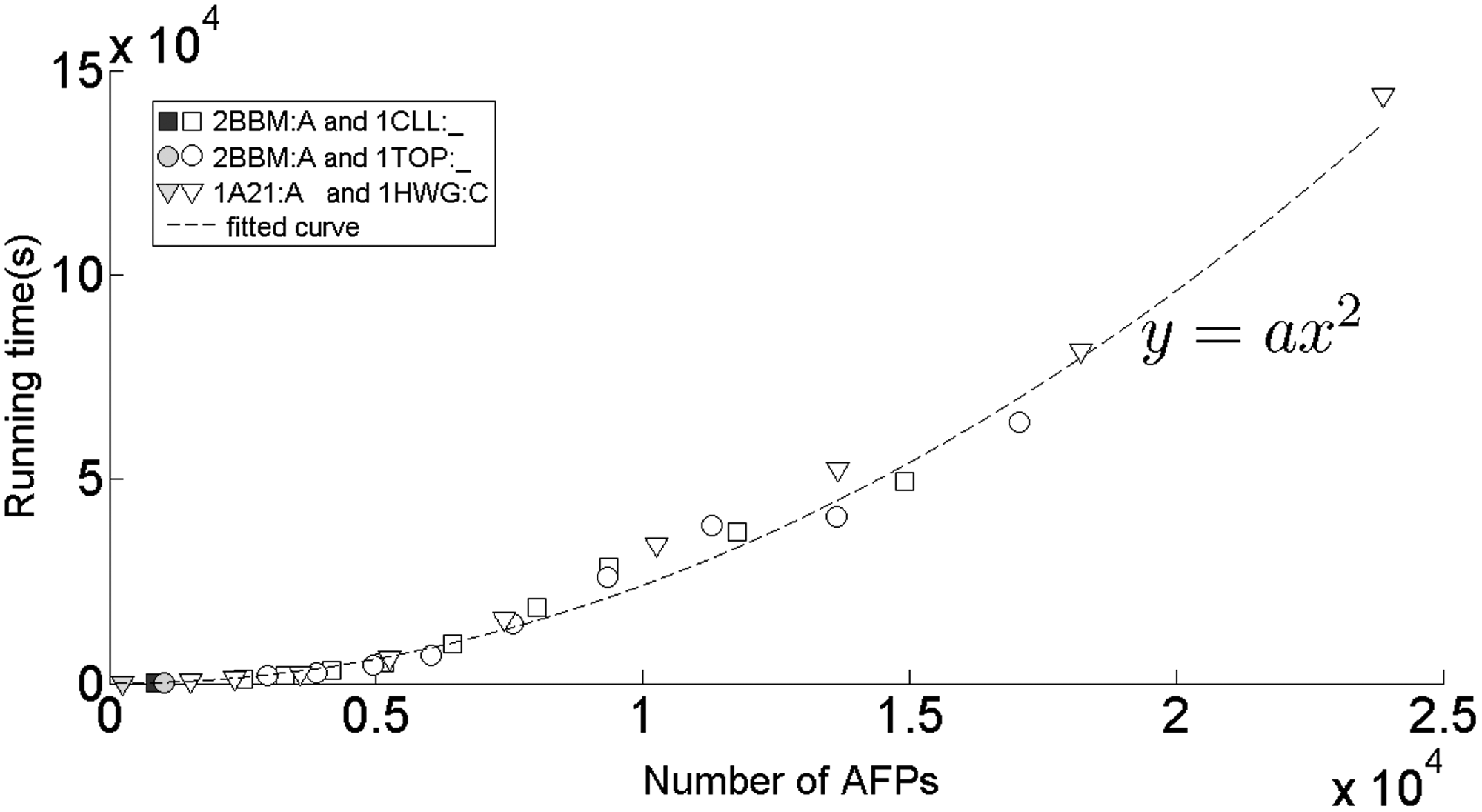
The relationship between the running time and the number of AFPs for three protein pairs: 2BBM:A and 1CLL:_, 2BBM:A and 1TOP:_, and 1A21:A and 1HWG:C. The unfilled marks represent the data produced using the constant-length AFPs. For each pair of protein, nine different numbers of AFPs are produced by setting the length of AFP to nine different values: 6, 7, 8, 9, 10, 11, 12, 13, and 14. The filled marks represent the data produced by FlexVAFP. All the data can be approximately fitted by a single curve: y = ax2, where a ≈2.3 × 10−4.
It can be seen from Figure 4 that the running time is approximately proportional to the square of the number of AFPs generated by the methods. So, the number of AFPs generated by an AFP-based method can reflect its efficiency. As seen from Table 5, it is clear that the number of AFPs produced by FATCAT is 10 times greater than that of FlexVAFP, which indicates that the running time of FlexVAFP is 100 times less than that of FATCAT. The use of the variable-length AFPs in FlexVAFP reduces the number of AFPs, which greatly improves the efficiency of the structure alignment.
4. Conclusion
In this article, we have developed a new method to identify the local similar fragments between protein structures and to obtain the optimal structure alignment results by introducing as few twists as possible, which is realized by dynamic programming with an improved transition function. The proposed method identifies the local similar fragment pairs (called AFPs) of protein structures by defining a local coordinate system of a Cα atom using three previous consecutive residues, which simplifies the computation at each step of the expansion of AFPs. Our method also allows different AFPs to have different lengths, which can greatly reduce the number of AFPs and improve the efficiency of the proposed method compared with other methods that use constant-length AFPs. In addition, the FlexVAFP method allows nonconsecutive AFPs to share the same transformation during the concatenation, which helps in reducing the number of twists in the structure alignment. Compared with other methods, the proposed structure alignment method can produce competitive results with fewer twists and shorter running time.
Footnotes
Acknowledgments
This work is supported by the National Science Foundation of China (Grant no. 61272213) and the Fundamental Research Funds for Central Universities (Grant no. lzujbky-2016-k07). The authors would also like to thank anonymous reviewers for their valuable comments.
Author Disclosure Statement
No competing financial interests exist.