
Abstract
Abstract
The cellulases from different cellulolytic organisms have evolutionary relationships, which range from single-celled prokaryotes to the complex eukaryotes of the living world. This in silico analysis revealed the presence of a conserved cellulase domain along with evolutionary relationships among cellulases from several species of Archaea, Bacteria, and Eukarya. The amino acid sequences of cellulases from Archaea and Bacteria showed closer identity with their domain or phylum members that provided insights into convergent and divergent evolution of cellulases from other enzymes with different substrate specificities. Evolutionary relatedness was also observed in phylogenetic trees among a number of cellulase sequences of diverse taxa. In cellulases, propensity for alanine, glycine, leucine, serine, and threonine was high, but low for cysteine, histidine, and methionine. Catalytic aspartic acid had a higher propensity than glutamic acid, and both were involved in regular expression patterns. Characteristic group and multigroup-specific conserved signature indels located in the catalytic domains of cellulases were observed that further clarified evolutionary relationships. These indels can be distinctive molecular tools for understanding phylogeny and identification of unknown cellulolytic species of common evolutionary descent in different environments.
1. Introduction
I
The Carbohydrate-Active enZYmes (CAZy) database provides information about the families of structurally related catalytic and noncatalytic (carbohydrate binding) domains of enzymes that catalyze the biosynthesis, breakdown, or modification of carbohydrates and glycoconjugates (Lombard et al., 2014). In the CAZy database, cellulases are grouped into the GH families, which include endoglucanase (GH 5, 6, 7, 8, 9, 10, 12, 26, 44, 45, 48, 51,74, and 124), cellobiohydrolase (reducing end; GH 7, 9 and 48), cellobiohydrolase (nonreducing end; GH 5, 6, and 9), and β-glucosidase (GH 1, 2, 3, 5, 9, 30, and 116), whereas some are yet to be assigned to families. Cellulases have widespread industrial applications in food, brewery and wine, animal feed, textile and laundry, pulp and paper industries, and in agriculture and research purposes (Bhat, 2000; Phitsuwan et al., 2013). The growing demand for cellulases because of their potential for the production of renewable liquid fuels, as alternatives to fossil fuels, from biomass sugars, has led to the exploration of cellulolytic organisms in a wide range of sources (Wilson, 2009; Thomas et al., 2016).
Microorganisms have the ability to exist in a wide range of habitats with distinct environmental restrictions (Ram et al., 2014). Owing to the abundant availability of the organic cellulose, myriads of diverse cellulolytic organisms are contemplated to be present in nature, for which probes based on cellulase enzymes could be highly helpful for their evaluation (Eveleigh et al., 1995; Sukharnikov et al., 2011). One such important probe is conserved signature indels (CSIs) generally of defined size and flanked by conserved regions in protein sequences, which provide a useful tool for understanding phylogenetic relationships (Gupta, 1998). Taxa-specific CSIs, exhibiting high predictive value and group specificity, are good phylogenetic markers and provide valuable information regarding the branching order and interrelationships among different taxon groups. Hence, based on their presence or absence, it becomes possible to identify both known and even previously unknown species of common evolutionary descent in different environments (Gupta and Griffiths, 2002). Presently, the evolutionary relationships among cellulolytic organisms are not well known and there are no distinctive CSIs reported for cellulase enzymes and, thus, their knowledge can be useful for exploring the diversity and evolutionary relationships of cellulolytic microbes.
In this study, amino acid sequences of the cellulases of Archaea and Bacteria were used to determine the phylogenetic relationships among cellulolytic organisms. Many CSIs, consisting of distinct inserts and deletions (indels), were identified in cellulases present in widely distributed taxa. These signatures also clarified the evolutionary relationships between members of different taxa.
2. Materials and Methods
2.1. Cellulase sequences
Amino acid sequences of cellulases including endoglucanase (EC 3.2.1.4), cellobiohydrolase (EC 3.2.1.91), and β-glucosidase (EC 3.2.1.21) from species belonging to various phyla of domains Archaea and Bacteria were used as query to assess their evolutionary neighbors from the available UniProtKB and NCBI databases (Table 1).
ENZYME entry EC 3.2.1.4 (endoglucanase), EC 3.2.1.91 (cellobiohydrolase), EC 3.2.1.21 (beta-glucosidase).
GH, glycoside hydrolase.
2.2. Retrieval of reference sequences
The amino acid sequences of each cellulase query were used to BLAST in the UniProtKB database, with parameters E-threshold value cutoff (0.01), matrix (BLOSUM 62), filtering (none), gapped (yes), and hits (500). The validity of obtained references was further substantiated by a position-specific iterated BLAST (PSI–BLAST) algorithm of the NCBI, with parameters expect threshold cutoff (0.01), matrix (BLOSUM 62), gap costs (existence: 11 extension: 1), compositional adjustments (conditional compositional score matrix adjustment), filters and masking (none), and PSI–BLAST threshold cutoff (0.005) (Altschul et al., 1997). This resulted in 500 reference sequences, which were sorted according to their descending order of identity with query. These sequences were retrieved in FASTA format for their alignment.
2.3. Detection of CSIs
Fragmented, hypothetical, redundant, and uncharacterized sequences were deleted from the retrieved reference amino acid sequences. The remaining nonredundant set of reference sequences were aligned by MUSCLE in MEGA6 (Edgar, 2004). The taxonomic position of references, frequencies of taxa represented among reference sequences, and conserved patterns of amino acids were analyzed. The aligned sequences were examined for the presence of CSIs with flanked identical/conserved regions of sequences (Gupta, 2016). The location of indels in cellulases was determined by the search of indel containing amino acid sequences of individual references in the conserved domain database of the NCBI.
2.4. Evolutionary relationships among cellulolytic taxa
The evolutionary relationships of cellulases were inferred using the neighbor-joining method with 1000 bootstrap replications and were in the units of number of amino acid substitutions per site (Felsenstein, 1985; Saitou and Nei, 1987). This was corroborated by the Jones–Taylor–Thornton matrix-based model, in which the rate variation among sites was modeled with a gamma distribution (shape parameter = 1) along with complete deletion of gaps and homogeneous pattern among lineages (Jones et al., 1992). Evolutionary analyses were conducted in MEGA6 (Tamura et al., 2013).
2.5. Amino acid propensity
The propensity (total frequency of occurrence) of different amino acid residues among reference cellulases was determined in MEGA6. Among all queries, average frequencies of amino acid residues and standard deviations were calculated using Microsoft Excel 2007.
3. Results
3.1. Identities of cellulase queries and frequency of taxa distribution among cellulase references
It was observed that most cellulase queries had highest identities with the cellulases of the species of its domain (for Archaea) or phylum (within domain Bacteria) (Supplementary Table S1). Moreover, identities were also observed with other cellulases and functionally different enzymes. However, four queries in domain Bacteria including Treponema caldarium (Spirochaete), Sulfurihydrogenibium azorense (Aquificae), Fibrobacter succinogenes (Fibrobacteres), and Cellulomonas fimi (Actinobacteria) had highest identities to members of different phyla. Among reference sequences, the observed frequency of distributed taxa varied for each query, thus revealing differential cellulase identities of each query with diverse taxa (Supplementary Table S2). Within the domain Bacteria, there were 22 phyla, and in the domain Eukarya, there were 19 groups including 15 phyla, 1 subphylum (Tunicata), 2 divisions (Rhodophyta and Chlorophyta), and 1 Streptophyta (an unranked clade of the kingdom Plantae). However, major representations within the domain Bacteria were that of phyla Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes, whereas within Eukarya, they were that of Arthropoda, Nematoda, and Fungi (Ascomycota and Basidiomycota).
Unclassified taxa (Caldithrix abyssi, Candidatus Saccharibacteria, Gemmatimonadetes bacterium, Microgenomates, Parcubacteria, Peregrinibacteria, and Thermobaculum terrenum) were present among reference cellulases of several queries. Intriguingly, virus represented by the bacteriophage of Erwinia sp. (Erwinia phage of Caudovirales) was found in cellulase references of Staphylothermus hellenicus and Pyrococcus abyssi.
3.2. Regular expression patterns of catalytic amino acid residues
Several amino acid residues in cellulases of reference sequences were conserved between distantly related taxa. At certain positions in the alignment, amino acid residues of similar physiological properties were present and would substitute each other in different references, indicating their position-specific importance. Such residues were polar/hydrophilic [asparagine (N), glutamine (Q), serine (S), threonine (T), lysine (K), arginine (R), histidine (H), aspartic acid (D), and glutamic acid (E)] and nonpolar/hydrophobic [alanine (A), valine (V), leucine (L), isoleucine (I), proline (P), tyrosine (Y), phenylalanine (F), tryptophan (W), methionine (M), and cysteine (C)]. Furthermore, serine–threonine–proline (S–T–P) containing regions were observed mostly at the N terminus of different bacterial and fungal references.
Among the reference sequences for each query, two amino acid residues aspartic acid (D) or glutamic acid (E) that serve either as the nucleophile or proton donor in the active site for cellulose hydrolysis were found to be conserved. These catalytic residues were regularly surrounded by certain amino acids in a conserved manner and had regular expression patterns, which were furthermore similar for several cellulase queries (Table 2). When more than three physiologically different residues were present at one position in aligned sequences, it was denoted as X (for any amino acid) in regular expression patterns (Table 2). For instance, among references for Pyrococcus abyssi, proton donor E had the regular expression pattern D[ILV]X[NS]EP[HX], where [ILV] indicates I or L or V at this position, X indicates any amino acid, [NS] indicates N or S at this position, and [HX] indicates H in majority of references but were also substituted by other amino acids at this position in certain species. One particular pattern for endoglucanase and β-glucosidase was observed, in which residues N and P mostly surrounded the proton donor E, while for exoglucanase, nucleophile D had the regular expression pattern G[EX][SCX]DG.
Not known.
Regular expression not consistent among references.
3.3. Evolutionary relationships among cellulolytic taxa
In the phylogenetic trees, in majority, an overall taxa (Archaea, Bacteria, and Eukarya)-specific clustering of cellulase references was observed for all queries, therefore, indicating an evolutionary relatedness (Fig. 1, Supplementary Figs. S1–S15). However, certain members of different phyla including cellulases and other enzymes also indicated closer relationships with phylogenetic trees. Among Staphylothermus hellenicus references, members of different phyla that showed relationships are Paenibacillus macerans (Firmicutes) and Streptomyces rapamycinicus (Actinobacteria); Erwinia phage (Virus) and Rhizobium leguminosarum bv. trifolii (Proteobacteria); Mahella australiensis (Firmicutes) and Spirochaeta thermophila (Spirochaete); branch of Ktedonobacter racemifer (Chloroflexi) between members of Actinobacteria (Catenulispora acidiphila, Kitasatospora cheerisanensis, Acidothermus cellulolyticus, Amycolatopsis mediterranei, Actinotalea fermentans, Lechevalieria aerocolonigenes, and Micromonospora carbonacea); monophyletic branching of Mycobacterium vanbaalenii (Actinobacteria) with Proteobacteria members (Azorhizobium caulinodans, Skermanella stibiiresistens, and Acetobacteraceae bacterium); Herpetosiphon aurantiacus (Chloroflexi) shared a common ancestor with Proteobacteria members (Agarivorans albus, Shewanella violacea, and Teredinibacter turnerae) (Fig. 1). Such relationships were also observed among references of other cellulase queries (Supplementary Figs. S1–S15).
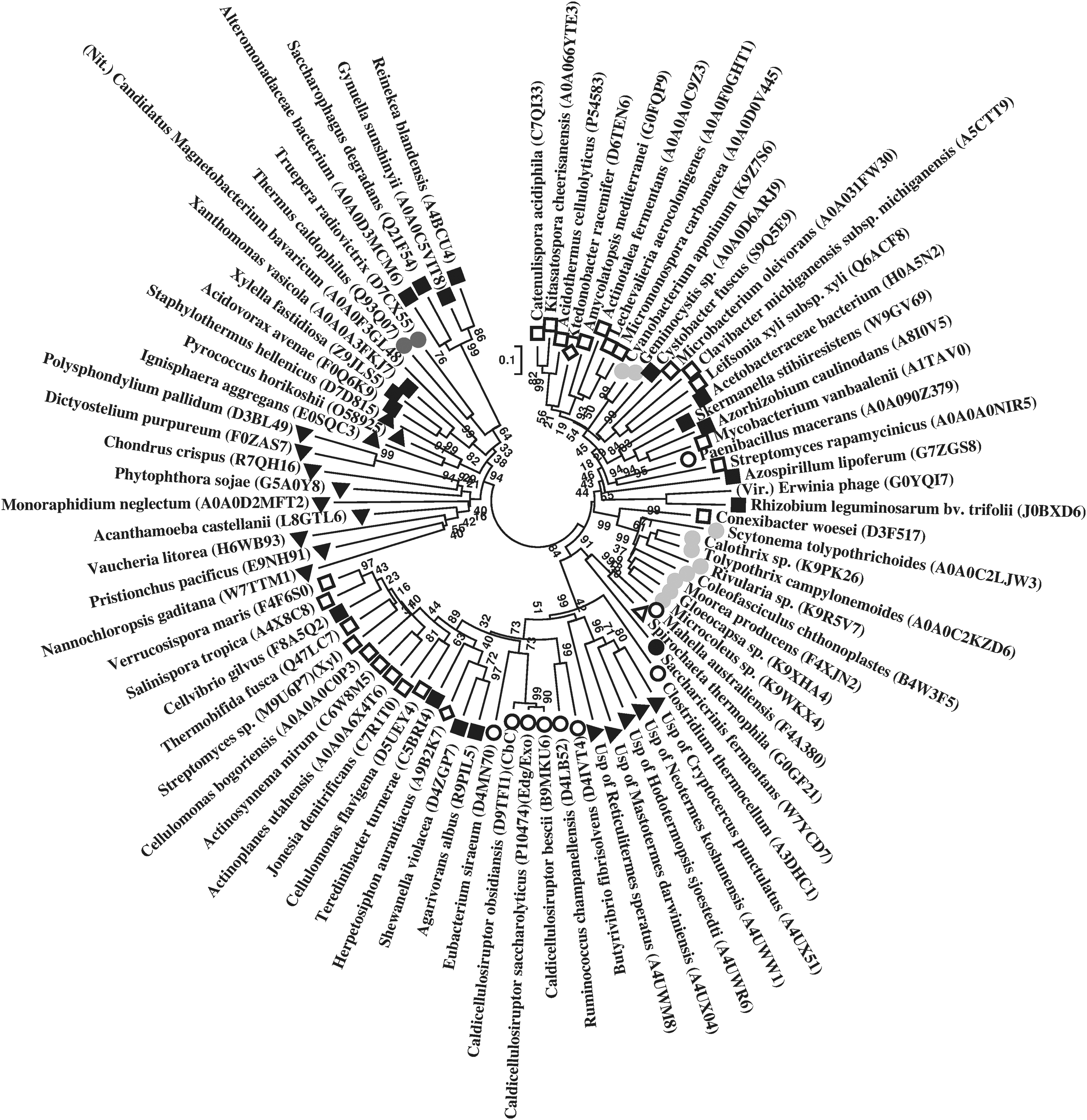
Phylogenetic tree of Staphylothermus hellenicus cellulase (endoglucanase; EC 3.2.1.4; GH 5) references, based on their amino acid sequences (accession numbers in parentheses). Symbols and abbreviations—Archaea (inverted closed triangle); Bacteria: Actinobacteria (open square), Bacteroidetes (closed circle), Firmicutes (open circle), Chloroflexi (open diamond), Cyanobacteria (light grey circle), Deinococcus-Thermus (dark grey circle), Nitrospirae (Nit.), Proteobacteria (closed square), Spirochaetes (open triangle); Eukarya (closed triangle), uncultured symbiont protist (Usp); Virus (Vir.); Enzymes: cellulose-1,4-beta cellobiosidase (CbC), endoglucanase/exoglucanase (Edg/Exo), endo-1,4-beta xylanase (Xyl). Bar, 0.1 substitutions per nucleotide position. GH, glycoside hydrolase.
3.4. Taxa-specific CSIs
Within the alignment of reference sequences for each cellulase query, CSIs comprising inserts and deletions located in the catalytic domains were detected. Several CSIs present either in members of one phylum or species (group-specific) or shared between members of different phyla (multigroup-specific) were observed among cellulase references (Figs. 2–5; Supplementary Figs. S16–S18; Supplementary Tables S3–S11). Among these, several CSIs were shared at same alignment sites between various references (of the domains Archaea, Bacteria, and Eukarya) of cellulase queries, between different types of cellulases, and also between cellulases and other enzymes with different substrate specificities.

Multigroup-specific conserved signature indels (CSIs) (double brackets).

Multigroup-specific CSIs (double brackets).


Group-specific CSIs (double brackets).
3.5. Amino acid propensity
Overall in cellulases, highest propensities were observed for alanine, glycine, leucine, serine, and threonine, whereas cysteine, histidine, and methionine had lowest propensities (Supplementary Table S12). Among catalytic residues, aspartic acid had a higher propensity than glutamic acid.
4. Discussion
This analysis revealed evolutionary relationships among widely distributed cellulases of organisms from diverse environments. Cellulases from the members of domains Archaea, Bacteria, and Eukarya showed homology at structural as well as physiological levels, which also indicates the conserved hydrolytic mechanisms among cellulases. Such a broad distribution of cellulolytic capability is highly indicative of a convergent evolution of cellulases under the selective pressure of the abundant availability of cellulose, which had occurred subsequently when the biosynthesis of cellulose had evolved with the emergence of algae, land plants, and other cellulose polymer-producing organisms (Lynd et al., 2002). The majority of cellulase queries had a high identity with cellulases of species that shared phylum with them, which supported the evolutionary relatedness among them. Their relatedness was also indicated by the taxa-specific clustering of cellulase references throughout the phylogenetic trees and by the sharing of CSIs among these members.
The cellulase queries showed both identities and formed clades in phylogenetic trees with other types of cellulases, which are all involved in hydrolyzing β-1,4-glycosidic linkages in different cellulosic substrates. Overall, endoglucanase (EC 3.2.1.4) queries had identities with bifunctional endoglucanase/exoglucanase, cellulose-1,4-beta-cellobiosidase (EC 3.2.1.91), glucosamine-link cellobiase (EC 3.2.1.21), cellodextrinase (EC 3.2.1.74), endo-1,3(4)-beta-glucanase (EC 3.2.1.6), and endo-1,3-beta-glucanase (EC 3.2.1.39). The endoglucanase (GH 45) of Cellvibrio japonicus had identities with other endoglucanases of Cellvibrio japonicus that belonged to different GH families, including endoglucanase C (GH 5) and endoglucanase A (GH 9) (Supplementary Table S1). Cellobiohydrolase (EC 3.2.1.91) in majority had a high identity with bifunctional beta 1,4-endoglucanase/cellobiohydrolase, whereas β-glucosidase (EC 3.2.1.21) had identity with 1,4-beta-
Several CSIs were also shared among these cellulases, including a four amino acid (aa) long insert between cellobiohydrolase and endoglucanase (Supplementary Table S3), a 3 aa long insert between endo-1,3-beta-glucanase and endoglucanase (Fig. 4a; Supplementary Table S6), 4 aa and 20 aa long inserts (Supplementary Fig. S16a) and a 2 aa long insert (Supplementary Fig. S17a) between bifunctional beta 1,4-endoglucanase/cellobiohydrolase and cellobiohydrolase, a 2 aa long insert between cellulose-1,4-beta-cellobiosidase, endoglucanase/exoglucanase, and endoglucanase (Supplementary Fig. S16b), and a 2 aa long insert between bifunctional beta 1,4-endoglucanase/cellobiohydrolase and endoglucanase (Supplementary Table S10). Such identities among different cellulase enzymes suggest a high degree of convergent evolution toward the hydrolysis of β-1,4-glycosidic linkages, but with different-sized cellulosic substrate accommodating structural differences. Furthermore, these multiple enzyme components of cellulases are considered to be required to cope with the physical diversity of cellulosic substrates (Béguin and Aubert, 1994). Microorganisms are known to produce complex combinations of cellulases, hemicellulases, and pectinases to completely utilize natural cellulose.
The cellulase queries also showed identities and formed clades in phylogenetic trees with functionally different enzymes, including various hydrolases [endo-1,4-beta-xylanase (EC 3.2.1.8), chitinase (EC 3.2.1.14), beta-galactosidase (EC 3.2.1.23), beta-mannosidase (EC 3.2.1.25), beta-xylosidase (EC 3.2.1.37), beta-fucosidase (EC 3.2.1.38), chitobiase (EC 3.2.1.52), alpha-
Inclusion of Eukarya (Arthropoda, Nematoda, Protist, Ciliophora, Rotifera, Echinodermata, and Cnidaria) references was observed for various cellulase queries. This most likely could be attributed to the involvement of horizontal gene transfer events from fungal or prokaryotic donors (Steele et al., 2004; Mitreva et al., 2009; Tanimura et al., 2013; Szydlowski et al., 2015). This was also supported by the presence of a one aa insert in endoglucanase of Arthropoda and Fungi references of Cellvibrio japonicus cellulase (Fig. 3a) and of two aa inserts shared between uncultured symbiont protists and different bacterial phyla members (Supplementary Fig. S16b). Among Dickeya dadantii references, branching of a Nematoda member (Aphelenchus avenae) was observed between members of Bacteroidetes in the phylogenetic tree (Supplementary Fig. S11), whereas another nematode Pratylenchus goodeyi shared a two aa insert with members of Bacteroidetes (Fig. 3b; Supplementary Table S6). This might be attributed to the mutualistic association of bacteria including Bacteroidetes with several plant-parasitic nematodes, which have been known to facilitate the nematodes to kill their host or to promote their development and propagation (Noel and Atibalentja, 2006; Tian et al., 2011). Cellulases of some Eukarya members including Mollusca, Tunicata, Arthropoda (like termites), Annelida, and Amoebozoa are even considered to have a separate origin (Monk, 1976; Davison and Blaxter, 2005).
Interestingly, references of Streptophyta that included land plants, were present among the cellulase queries of Salinarchaeum sp. Harcht-Bsk 1, Fibrobacter succinogenes, and Caldivirga maquilingensis. An insert of almost 13 aa specific to plants (Viridiplantae) was also observed (Fig. 4b) among the references of Fibrobacter succinogenes cellulase query. Plants are known to express cellulases for primary roles in cell growth, repairing or arranging cellulose microfibrils during cellulose biosynthesis (Hayashi et al., 2005), and abscission (Abeles, 1969).
The phylogenetic distribution of enzymes involved in cellulose utilization and synthesis at different taxonomic levels has been identified through analysis of several sequenced bacterial genomes (Berlemont and Martiny, 2013). In this study, evolutionary relationships observed for certain queries and for various cellulolytic references in phylogenetic trees were in consistent support of the reports of their proposed phylogenetic positions. Members of Spirochaete had a greater closeness to the members of Firmicutes. This was evident in the references obtained for Treponema caldarium cellulase, which showed maximum identity with the members of Firmicutes (Supplementary Table S1). Even in phylogenetic trees, Spirochaete members consistently formed a clade either with or branched between Firmicutes members. Moreover, CSIs of three aa long inserts in endoglucanase were also shared in these two phyla (Fig. 4a; Supplementary Table S10). Their relatedness has been suggested to the possible involvement of a common ancestor or of an interphyla horizontal gene transfer event (Caro-Quintero et al., 2012). Another cellulase query, Sulfurihydrogenibium azorense of Aquificae, showed maximum identity with the members of Proteobacteria (Supplementary Table S1), which even had the highest frequency of 89.8% among its references (Supplementary Table S2). The closer relationship of Aquificae with Proteobacteria has also been previously suggested (Cavalier-Smith, 2002), and they are also known to share a unique two aa long CSIs in the inorganic pyrophosphatase (Griffiths and Gupta, 2004).
Presently, Fibrobacteres, which consists of a single genus Fibrobacter, is recognized as a distinct phylum. The cellulase query Fibrobacter succinogenes showed maximum identity to the cellulases of Bacteroidetes, with which it furthermore either formed a clade (with Sporocytophaga myxococcoides, Supplementary Fig. S5) or shared a common ancestor (Supplementary Fig. S11) in phylogenetic trees. Similarly, phylogenetic studies based on Rpo C and Gyrase B protein sequences have also indicated that Fibrobacter succinogenes is closely related to Bacteroidetes, and hence they are also proposed to be part of a single superphylum (Gupta, 2004; Gupta and Lorenzini, 2007). Members of Actinobacteria including Mycobacterium spp. (M. rhodesiae and M. vanbaalenii) in majority of the phylogenetic trees had monophyletic grouping with the members of the phylum Proteobacteria (Fig. 1; Supplementary Figs. S2, S10, S13). Their closer relationship has been suggested to be because of either an ancient transfer of genes or a deep paralogy followed by retention of the genes (Kinsella et al., 2003). Actinobacteria cellulolytic members, Cellulomonas spp. (C. cellasea and C. fimi), in particular, also formed a clade in several phylogenetic trees with the Proteobacteria member Cellvibrio gilvus (Supplementary Figs. S3, S5, S6).
This observed relationship is also in agreement with the earlier report that Cellvibrio gilvus belongs to the genus Cellulomonas, which was based on their phylogenetic and whole-genome comparisons (Christopherson et al., 2013). Lentisphaera araneosa, a member of the phylum Lentisphaerae, formed a clade with Verrucomicrobiae of the Verrucomicrobia phylum (Supplementary Fig. S12). This supports the earlier considered view of their relationship as sister phyla based on 16S rRNA phylogenetic analysis (Cho et al., 2004). Fusobacteria member Fusobacterium mortiferum formed a clade with the Firmicutes member Clostridium cellulovorans (Supplementary Fig. S3). This is in agreement with the earlier report of its remote relatedness to Firmicutes, which was based on its comparative genomic analysis (Mira et al., 2004). Dictyoglomus thermophilum of the Dictyoglomi was found among cellulase references of queries Thermotoga maritima, Thermobispora bispora, and Leeuwenhoekiella blandensis. In all these, it had a common ancestor with Firmicutes. Among references of Thermotoga maritima, it branched between Firmicutes members, which had a common ancestor with the Thermotogae members (Supplementary Fig. S12). Both genome and orthologous protein sequence comparisons have also indicated that Dictyoglomus is most closely related to the phylum Thermotogae and that it forms monophyletic groups with certain members of Firmicutes (Conners et al., 2006; Nishida et al., 2011). Phaeodactylibacter xiamenensis of Bacteroidetes formed a clade with the Cyanobacteria member Aphanocapsa montana (Supplementary Fig. S3). This might be attributed to the fact that Phaeodactylibacter xiamenensis is among the bacteria that are found in the phycospheres of various microalgae, and may possess functional genes for acquiring nutrition from the host algae (Chen et al., 2014).
However, members of Chloroflexi exhibited phylogenetic positions with members of Actinobacteria, Cyanobacteria, Proteobacteria, and Firmicutes, which was also indicated by their sharing of several CSIs of 4 aa and 23 aa inserts (Fig. 2a), 4 aa and 20 aa inserts (Supplementary Fig. S16a), and a 2 aa insert (Supplementary Fig. S17a). Among the unclassified members, Thermobaculum terrenum formed a clade with the Chloroflexi member Roseiflexus castenholzii (Supplementary Figs. S2, S12). This supports the proposed placement of Thermobaculum terrenum in Chloroflexi, as it was also found to share three unique gene arrangements in their genomes (Kunisawa, 2011). Another unclassified member, Caldithrix abyssi, formed a clade with the Ignavibacteriae member Melioribacter roseus (Supplementary Fig. S3). Their closeness has also been indicated from the whole proteome analysis of Melioribacter roseus (Kadnikov et al., 2013).
The CSIs that are restricted to a particular clade or group of species provide useful phylogenetic markers for analyzing the taxa of common evolutionary descent (Gupta, 1998). Besides various multigroup CSIs, numerous unique group-specific CSIs were also observed among the members of Actinobacteria (Fig. 5a, b; Supplementary Tables S4, S8), Proteobacteria (Supplementary Table S9), Fungi (Supplementary Table S11), and Viridiplantae (Fig. 4b). Other group-specific CSIs of five aa inserts were found exclusively among the species of Streptomyces of Actinobacteria (Supplementary Fig. S18a, b). Indel characters are considered less homoplasious that provide explicit phylogenetic signals (Rokas and Holland, 2000). From the high degree of specificity and location of the CSIs in the catalytic domains of cellulases, it is plausible that the genetic changes represented by these indels might be functionally important for the organisms possessing them.
A number of amino acid residues (histidine, cysteine, aspartic acid, glutamic acid, arginine, lysine, tyrosine, serine, threonine, asparagine, glutamine, and tryptophan) are known to be involved in different enzymatic catalysis (Holliday et al., 2009). Among various amino acid residues, alanine, glycine, leucine, serine, and threonine formed a major part of cellulases. As aspartic acid and glutamic acid are the known catalytic residues for cellulases, their propensities were also comparatively higher than the other catalytic residues except serine and threonine. However, serine and threonine are not involved in catalysis, but are mostly, in addition to proline (in the form of S–T–P), enriched in the linker regions that separate catalytic and carbohydrate binding modules in cellulases. Here, extensive O-linked glycosylation is known to occur, which suggests that cellulase linkers exhibit functions beyond simple domain connectivity (Sammond et al., 2012).
Footnotes
Acknowledgments
Fellowships from the Council of Scientific and Industrial Research (CSIR, New Delhi) to Lebin Thomas, from the University Grants Commission (UGC, New Delhi) to Hari Ram, and the DST-purse and R&D grants from the University of Delhi are gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.