
Abstract
Abstract
This article examines three techniques for rapidly assessing the electrostatic contribution of individual amino acids to the stability of protein–protein complexes. Whereas the energetic minimization of modeled oligomers may yield more accurate complexes, we examined the possibility that simple modeling may be sufficient to identify amino acids that add to or detract from electrostatic complementarity. The three methods evaluated were (a) the elimination of entire side chains (e.g., glycine scanning), (b) the elimination of the electrostatic contribution from the atoms of a side chain, called nullification, and (c) side chain structure prediction using SCWRL4. These techniques generate models in seconds, enabling large-scale mutational scanning. We evaluated these techniques on the SMAD2/SMAD4 heterotrimer, whose formation plays a crucial role in antitumor pathways. Many studies have documented the clinical and structural effect of specific mutations on trimer formation. Our results describe how glycine scanning yields more specific predictions, although nullification may be more sensitive, and how side chain structure prediction enables the identification of uncharged-to-charge mutations.
1. Introduction
E
Predicting the electrostatic influences of specific mutations is challenging, in part, because mutations simultaneously create both structural and electrostatic changes. Replacing a charged side chain with an uncharged side chain alters the electrostatic field and it can also alter the pattern of side chain packing and solvent exposure nearby. Changes in the solvent exposure of a charged group can locally decrease the dielectric of the polar solvent, enhancing nearby electrostatic potentials through electrostatic focusing (Klapper et al., 1986; Polticelli et al., 1999; Rohs et al., 2009; Blumenthal et al., 2013). Although the computationally intensive simulation of mutant structures could better approximate realistic packing, we hypothesized that computationally inexpensive modifications may be sufficient to approximate the effect of most mutations and thus rapidly screen for electrostatically influential amino acids.
This article evaluates three computationally inexpensive ways to modify protein structures for the purpose of evaluating the electrostatic influence of specific amino acids on protein–protein recognition. The first method is glycine scanning, in which the side chain of each amino acid is systematically removed without modifying the rest of the structure. The second method, which we call “nullification,” systematically eliminates the electrostatic contribution of each amino acid within each member of an oligomer. Nullification maintains the position of all atoms, ensuring that the spatial arrangement of dielectrics remains identical to the original structure and thus that the shielding influence of structure does not change with the field. Finally, the third approach applies the program SCWRL (Krivov et al., 2009) to predict the conformations of all 20 possible amino acids at significant positions along the backbone. The effect of each method on electrostatic complementarity was evaluated using VASP-E, a method we described earlier (Chen, 2014). Our comparison revealed several interesting observations about how these techniques can predict amino acids that have an electrostatic influence on binding and specificity.
We evaluated these predictors on the two heteromeric interfaces of the SMAD2–SMAD4 trimer (Chacko et al., 2004), which is part of a signaling pathway with antitumor effects (Xu et al., 2006; Hardwick et al., 2008; Zhang et al., 2010; Ding et al., 2011; Itatani et al., 2013; Hernanda et al., 2015). The formation of this trimer, from one SMAD4 and two SMAD2 subunits, is supported, in part, by strong electrostatic interactions across its heteromeric interfaces (Chacko et al., 2004). Mutations that eliminate certain charge–charge interactions are frequently associated with degradations of trimer formation and hereditary disease. Careful data gathering by researchers in the field [e.g., Wooderchak et al. (2010)] has revealed that many missense mutations of SMAD4 are experimentally associated with juvenile polyposis syndrome (JPS) (Howe et al., 1998; Schwenter et al., 2012), a hereditary disorder characterized by the formation of polyps in the rectum, colon, or gastrointestinal (GI) tract, and hereditary hemorrhagic telangiectasia (HHT) (Gallione et al., 2004, 2006), a different hereditary disorder that causes recurring nosebleeds, the dilation of capillaries in the mouth, face, hands, and GI tract, and venous malformation. We used charge-eliminating SMAD4 missense mutants as a benchmark to evaluate the accuracy of hypothetical mutation testing.
2. Methods
2.1. Measuring changes in electrostatic complementarity
We use VASP-E (Chen, 2014) to measure electrostatic complementarity. We paraphrase this technique here for convenience and to clarify the experimental design. Beginning with a complex of two proteins, A and B, we first prepare each structure by stripping existing hydrogens and modeling their positions using MolProbity (Chen et al., 2009). Next, we use Delphi (Honig and Nicholls, 1995; Rocchia et al., 2001) to solve the electrostatic potential field for each protein in isolation. We then use VASP-E to produce electrostatic isopotential surfaces at a given range of isopotential thresholds
The isopotentials generated by VASP-E are represented as three-dimensional geometric solids. Using Boolean set operations (Fig. 1), VASP-E can compute the intersection region where two or more isopotentials overlap. We denote Boolean intersections with

Boolean operations on geometric solids. Input surfaces are shown in gray with dotted lines. Output surfaces for three basic operations are shown in gray with solid lines.
To measure electrostatic complementarity between oligomers A and B at a given isopotential threshold a, we use VASP-E to compute Boolean intersections between
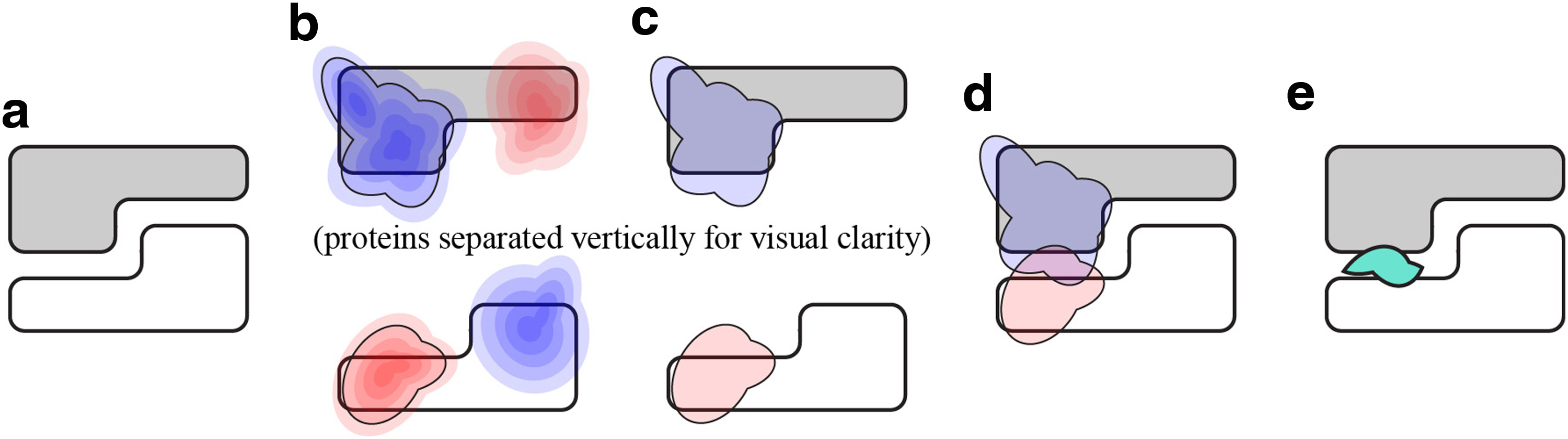
Computing electrostatic complementarity with Boolean intersections.
To evaluate the electrostatic effect of a modeled change on subunit A of complex AB, we first modify A to produce
Although changes in complementarity, measured in this way, can be predictors of amino acids that have an electrostatic influence on specificity (Chen, 2014), the magnitude of the difference in complementarity does not necessarily correlate with differences in binding affinity (Chong et al., 1998). Generally speaking, we find that after evaluating a range of electrostatic thresholds, one positive and one negative threshold are generally most informative, with other thresholds reiterating similar results.
When examining the contribution of individual amino acids, at a given isopotential threshold, we predict that an amino acid has an electrostatic influence on affinity when the difference in complementarity between the wild type complex and the modified complex is greater than one-half the maximum positive or the maximum negative difference observed for all amino acids at this isopotential. We refer to half the maximum positive difference or half maximum negative difference as the positive and negative prediction thresholds, respectively. This approach was applied in earlier methods, in which such differences in electrostatic isopotential were effective for distinguishing influential amino acids (Chen, 2014).
2.2. Creating electrostatic variations at the protein–protein interface
To evaluate techniques for examining the contribution of an amino acid on electrostatic complementarity, we considered three methods for generating electrostatic variations in the protein–protein interface. The first two methods, glycine scanning and nullification, eliminate the electrostatic contribution of individual amino acids. By detecting the removal of charged amino acids that add or detract from electrostatic complementarity, they yield insights into the effect of mutating charged amino acids into uncharged amino acids. The third method, using SCWRL for side chain structure prediction, both adds and removes charged amino acids. This approach adds the further capability of evaluating the effect of mutating uncharged amino acids into charged amino acids.
2.2.1. Glycine scanning
Glycine scanning modifies the input complex by removing the side chain from one amino acid in one protein of in the input complex. No compensation for the absence of this side chain is made: the changes in side chain packing and backbone conformation that inevitably occur are not modeled in this approach. After a side chain is removed, the complex is examined in the manner described in Section 2.1. This process is repeated for every amino acid in the oligomer. Finally, the difference in complementarity between the original complex and the modified complex is plotted for every amino acid.
2.2.2. Electrostatic nullification
Earlier, we developed electrostatic nullification as one technique to evaluate the influence of one amino acid in molecular interactions (Chen, 2014). This technique uses Delphi (Rocchia et al., 2001) to eliminate the electrostatic contribution of one amino acid in one of the proteins in the complex. Although the electrostatic contribution is eliminated, the atomic geometry of the amino acid remains fixed, thereby decoupling changes to the electrostatic field and the structure. The effect is that a region of space that was once occupied by the amino acid remains inaccessible to the solvent, creating a low-dielectric zone that would have reverted to the high-dielectric zone of the solvent if the amino acid were removed completely. Once the electrostatic contribution of the amino acid is eliminated and the electrostatic potential field is recomputed, we evaluate the change in electrostatic complementarity. This process is repeated for every amino acid in both proteins in the complex.
2.2.3. Side chain structure prediction
We use SCWRL (Krivov et al., 2009) to modify the input complex by replacing the side chain of one amino acid with the side chains of the remaining 19 standard amino acids. SCWRL selects the conformation of the side chain to avoid steric hindrance with the existing structure, but no compensation is made for changes at the protein–protein interface that may cause steric hindrance with the protein binding partner. For each substituted amino acid, the entire complex is examined in the manner described in Section 2.1. Only single mutants are considered at a user-selected range of amino acids to limit the computational cost of examining the huge of all possible single mutants. Once the electrostatic isopotentials are created, we plot the difference in complementarity between the original complex and the side chain-substituted complex.
2.3. SMAD2–SMAD4 trimer
Mothers against decapentaplegic homologue 4 (SMAD4) are involved in cell signaling by modulating members of the transforming growth factor B (TGF-B) protein superfamily (Chacko et al., 2004). SMAD4 forms heteromeric complexes with other SMAD homologues, such as SMAD2 and SMAD3 (Lagna et al., 1996). These complexes accumulate in the nucleus of a cell and function to regulate the transcription of many target genes by acting as transcription factors (Massagué et al., 2005). These intracellular mediators are important for transcriptional and antiproliferative responses to TGF-B. The signals that propagate through the SMAD/TGF-B pathway relate to cell growth, adhesion, migration, cell fate determination and differentiation, and apoptosis. As these signals are vital to the life of a cell, malfunctions in the SMAD/TGF-B pathway have been discovered in many human diseases, such as pancreatic cancer, head and neck cancers, and colorectal cancer, as well as juvenile polyposis and HHT (Gallione et al., 2004). More specifically, the SMAD4 protein has been found to be inactive in nearly half of pancreatic carcinomas (Hahn et al., 1996). Owing to the severity of these diseases, it is desirable to determine the source of these malfunctions.
Previous studies reveal that SMAD2 and SMAD4 form a trimer through a conserved protein–protein interface, and the majority of tumor-derived missense mutations map to this interface (Chacko et al., 2004). These mutations disrupt the formation of the trimer, indicating that the assembly of the SMAD4 is crucial for cell signaling and is disturbed by mutations that are ultimately tumorigenic. The crystal structure of the SMAD2–SMAD4 trimer (pdb: 1u7v), described earlier by Chacko et al. (2004), is illustrated in Figure 3. SMAD4, hereon referred to as the B subunit, forms two separate interfaces with the SMAD2 subunits, A and C, which share a separate interface. Together, the three subunits have no common interface, leaving an empty region in the center of the trimer. For this reason, we examined SMAD4 mutations on each interface independently of the third subunit.
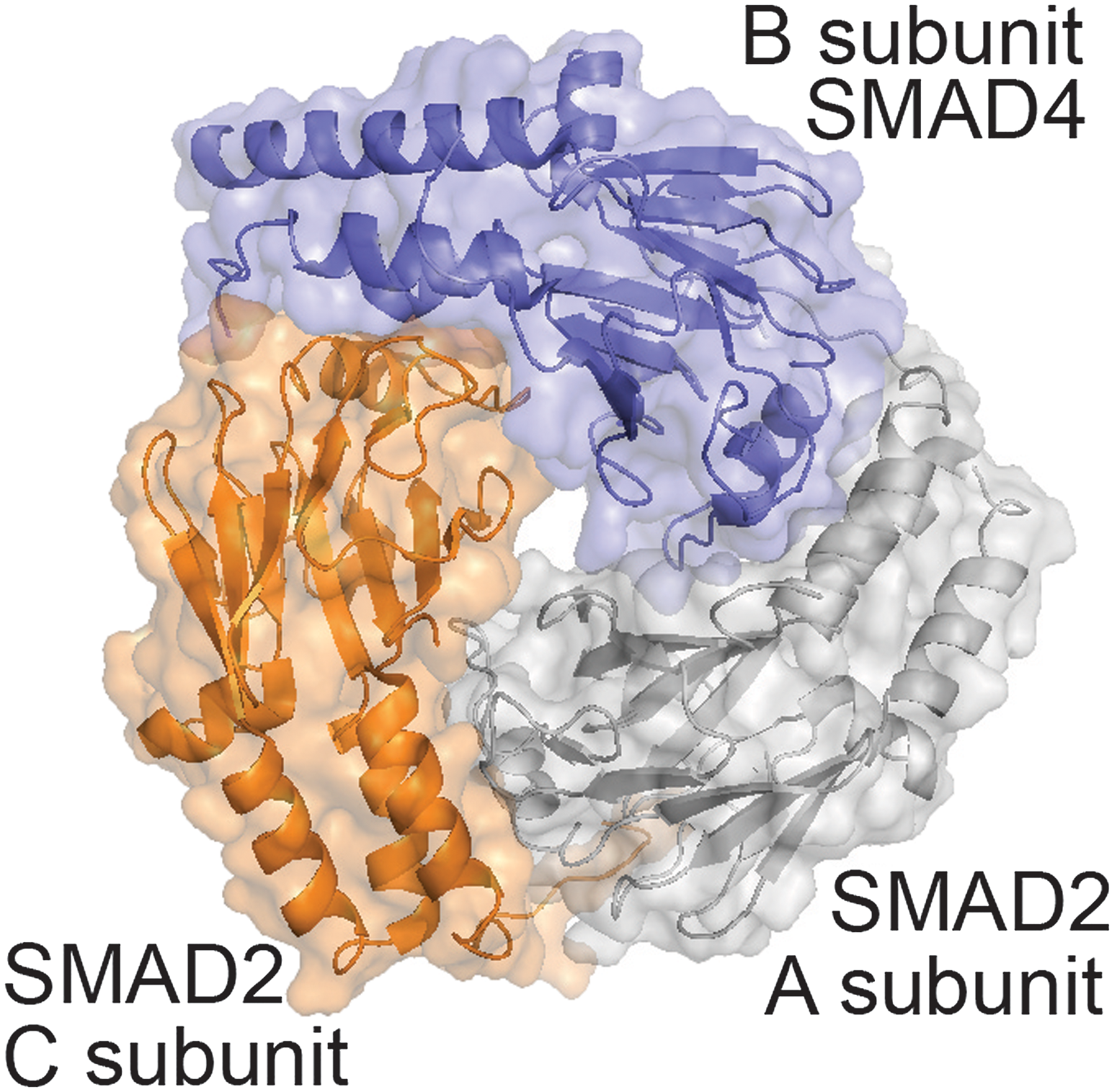
The SMAD2–SMAD4 trimer. The B subunit is shown in blue. SMAD2 subunits, A and C, are shown in orange and gray.
The Department of Pathology at the University of Utah offers a manually curated database, hereon referred to as the ARUP database, of many known clinically significant mutations in SMAD4 (Wooderchak et al., 2010). The mutations identified are associated with JPS and with HHT. To supplement the detailed literature search that populates the SMAD4 database, we performed our own literature search to identify mutations not mentioned. Altogether, these data form a data set of experimentally verified mutations that are associated with JPS and HHT. Using these data and other published literature as a gold standard, our computational results evaluate how accurately the three methods considered here can be used to predict a mutation that corresponds to one of these two conditions.
3. Results
We used the techniques already described to create structural variations of the SMAD2–SMAD4 trimer. Glycine scanning and nullification were used to predict charged residues that have an electrostatic influence on binding, by examining the effect of removing them. Side chain structure prediction was used to predict influential mutations at specific residue positions. All methods were evaluated against findings established in the experimental literature. We demonstrate the validity of our findings by explicitly citing the literature that supports our predictions. Results were, therefore, summarized in paragraph rather than in table form.
3.1. Glycine scanning and nullification on the AB interface
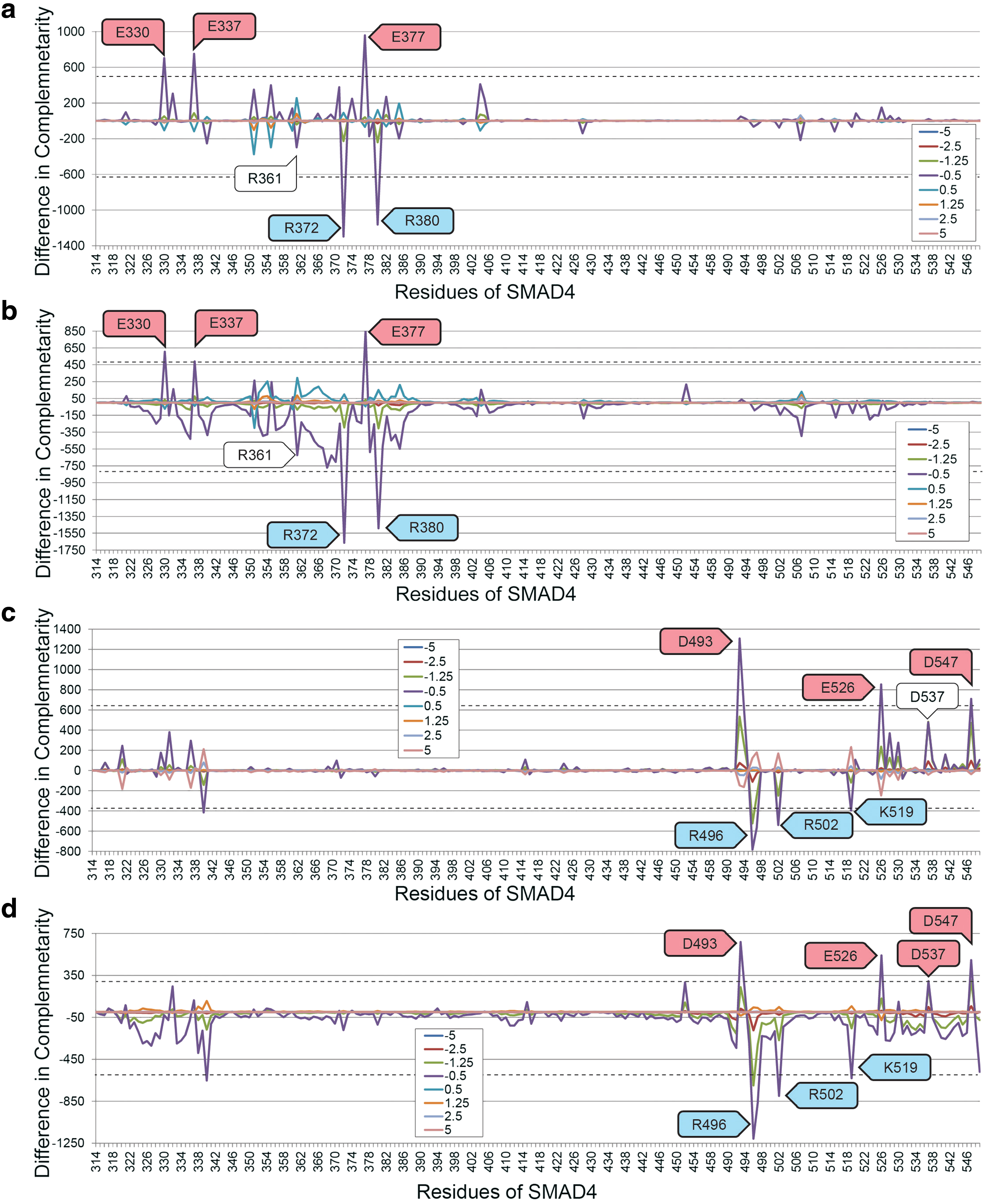
Glycine scanning, performed on the AB interface of the SMAD4 trimer, identified three negatively charged and two positively charged amino acids that surpassed the prediction threshold (Fig. 4a). These residues were E330, E337, R372, E377, and R380. All those identified play an experimentally established role in binding. Nullification detected the same amino acids (Fig. 4b).
Changes in electrostatic complementarity, in the BA
Mutations of E330 have been associated with JP in studies that observed the mutations E330G and E330K in patients and tumors (Sayed et al., 2002). E337 is known to form a charge–charge interaction with R243 on SMAD3 (Schiro et al., 2011). R372 and R380 are totally conserved in SMAD proteins and they form four and three hydrogen bonds, respectively, that stabilize the loop helix binding region of SMAD4 (Shi et al., 1997). E377 is also totally conserved in SMAD (Shi et al., 1997). The ARUP database associates several loss of charge mutations with R361, but glycine scanning and nullification of R361 did not create differences in electrostatic complementarity that were sufficient for detection.
3.2. Glycine scanning and nullification on the BC interface
Glycine scanning (Fig. 4c) and nullification (Fig. 4d) identified seven influential amino acids at the BC interface. These residues were D493, R496, R502, K519, E526, D537, and D547. All those identified play an experimentally established role in binding: D493 is frequently mutated to histidine in cancer (Hahn et al., 1996; Prokova et al., 2007).
Mutations of R496 to histidine likely disturb the formation of homo and heteromeric SMAD complexes (Chacko et al., 2001; Lazzereschi et al., 2005). Mutations of R502 disrupt the capacity for SMAD4 to activate the transcriptional response, thus disrupting its antitumor activity (Chacko et al., 2001). Loss of charge in K519 prevents monoubiquitylation of K519 and prevents complex formation with SMAD2 (Dupont et al., 2012). E526 forms intramolecular hydrogen bond networks that stabilize the binding interfaces of SMAD4 (Shi et al., 1997). D537 is mutated in colon cancer (Eppert et al., 1996; Shi et al., 1997), although K537 was less than the threshold in glycine scanning, but only slightly more than the threshold in nullification. Established evidence of mutations of D547 destabilizing the SMAD2–SMAD4 trimer could not be found.
3.3. Substitutions modeled with SCWRL
Using SCWRL, we modeled all 20 possible mutations of every cancer-related missense mutation identified by the ARUP database as well as the amino acids identified through glycine scanning and nullification. These results are illustrated in heat maps in Figure 5.

Changes in electrostatic complementarity, in the BA
The electrostatic effect of SMAD4 mutations modeled with SCWRL generally influenced electrostatic complementarity at only one interface. Two mutations, E390 and G510, did not have a substantial influence on electrostatic complementarity. Both amino acids lie distant from either of the SMAD2 subunits and lie buried in the SMAD4 core. The remaining mutations tested exhibited larger variations in electrostatic complementarity.
Loss of charge mutations caused notable changes in complementarity. Mutations to glycine, which partially duplicated the measurements from glycine scanning, affected complementarity less than opposite charge mutations, which had the greatest effect on complementarity in all cases. The ARUP database indicates that R361C, R361G, R361H, R361L, and R361S are all associated with hereditary disease, and all exhibited a similar disruption of electrostatic complementarity. Indeed, quite a few other mutations of R361, as predicted with SCWRL, appear to achieve the same outcome. Other mutations of R361 appear to achieve similar changes in electrostatic complementarity and may thus also degrade antitumor performance.
Gain-of-charge mutations exhibited changes in complementarity that corresponded to the identity and charge of the substituted side chain. Mutations to argenine, lysine, glutamate, aspartate, and histidine, performed on C363, Y353, G352, L364, G386, C324, W509, G491, L533, and W524, all had notable effects on complementarity. Electrostatic complementarity correctly predicted all gain-of-charge mutations associated by ARUP with hereditary disease.
We also evaluated mutations from uncharged amino acids to uncharged amino acids that are associated by ARUP with hereditary disease. Unsurprisingly, these amino acids were never predicted to have an electrostatic influence on affinity. Among the mutations Y353S, L364V, L364W, G491V, L533V, L533P, and W524L, no individual mutation altered electrostatic complementarity substantially from the wild type.
4. Discussion
We have performed a direct comparison between glycine scanning, nullification, and side chain structure prediction as tools for modeling the influence of mutation on affinity on the SMAD2–SMAD4 trimer. Overall, predictions made with glycine scanning and nullification were similar, both identifying several amino acids that have experimentally established roles in binding. Although small variations in electrostatic complementarity may have resulted in the prediction of slightly different amino acids, it is clear that both methods generally identify similar amino acids.
Differences in electrostatic complementarity in glycine scanning models generally tend toward 0 when an uncharged residue changes to glycine. In contrast, differences in electrostatic complementarity in nullification models often result in regions of electrostatic focusing, such as in the SMAD4 residues between 361 and 369. These amino acids have no net charge, but because their electrostatic contribution is totally eliminated. Because they continue to prevent the polar solvent from occupying the same space as the original amino acid, the effect is an enhancement in electrostatic potentials nearby. Thus, the effect of nullifying uncharged amino acids nearby a charged amino acid still changes electrostatic complementarity. This effect was not large enough in SMAD4 to create substantial inaccuracies. Glycine scanning does not exhibit this effect and should generally achieve superior prediction specificity, whereas nullification achieves greater prediction sensitivity.
Unlike glycine scanning and nullification, which only evaluate the loss of charged residues, side chain structure prediction can identify gain-of-charge mutations that alter electrostatic complementarity. In every case in which experimental data are available, gain-of-charge mutations were accurately predicted using SCWRL. Loss-of-charge and opposite charge mutations were identified as accurately as glycine scanning and nullification. Finally, given the exclusively electrostatic nature of the analysis performed, influential mutations of uncharged side chains to other uncharged side chains could not be distinguished from other mutations, as expected. Overall, these data indicate that glycine scanning has considerable potential for the rapid identification of potentially influential charged amino acids and that SCWRL provides an excellent tool for evaluating the effect of any mutation on electrostatic complementarity.
Footnotes
Acknowledgment
This work was supported, in part, by National Science Foundation Grant 1320137 to Brian Chen and Katya Scheinberg.
Authors' Contributions
B.E.N. and E.F. found the data set, implemented and automated the software, and performed the experiments. B.Y.C. conceived of the experiment and wrote the article.
Author Disclosure Statement
No competing financial interests exist.