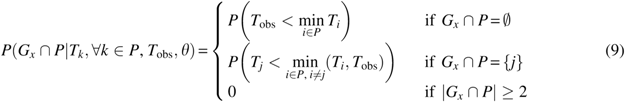High-throughput sequencing technologies have facilitated the generation of an unprecedented amount of genomic cancer data, opening the way to a more profound understanding of tumorigenesis. In this endeavor, two fundamental questions have emerged, namely (1) which alterations drive tumor progression and (2) in which order do they occur? Answering these questions is crucial for therapeutic decisions involving targeted agents. Because of interpatient heterogeneity, progression at the level of pathways is more reproducible than progression at the level of single genes. In this study, we introduce pathTiMEx, a generative probabilistic graphical model that describes tumor progression as a partially ordered set of mutually exclusive driver pathways. pathTiMEx employs a stochastic optimization procedure to jointly optimize the assignment of genes to pathways and the evolutionary order constraints among pathways. On real cancer data, pathTiMEx recapitulates previous knowledge on tumorigenesis, such as the temporal order among pathways which includeAPC,KRAS, andTP53in colorectal cancer, while also proposing new biological hypotheses, such as the existence of a single early causal event consisting of the amplification ofCDK4and the deletion ofCDKN2Ain glioblastoma.pathTiMEx is available as an R package.
1. Introduction
Over the last years, our basic understanding of tumorigenesis has increased substantially, primarily as a result of the large-scale use of high-throughput sequencing technologies. High-resolution genomic, epigenomic, transcriptomic, metabolomic and proteomic information from tens of thousands of cancerous samples is now publicly available (Cancer Genome Atlas Research Network, 2011; Cancer Genome Atlas Network et al., 2012; Cerami et al., 2012), providing the biomedical research community with an unprecedented amount of genomic data. In this context, two fundamental questions have emerged in describing tumorigenesis (Raphael and Vandin, 2014): (1) Which genomic alterations drive tumor progression? (2) What are the evolutionary constraints on the order in which these alterations occur? Answering these questions is crucial not only for therapeutic decisions but also for a basic understanding of carcinogenesis. Early genetic events, such as specific point mutations or copy number aberrations, have a particularly important role in tumor development as they may induce oncogenic addiction (Weinstein, 2002; Luo et al., 2009), a situation in which tumors rely on one single dominant oncogene for growth and survival (Torti and Trusolino, 2011). The identification of these events can prioritize candidate drug targets.
Alterations that provide a positive selective advantage and significantly contribute to tumor progression are termed drivers, while alterations that are selectively neutral are termed passengers. Intuitively, drivers and passengers can be classified according to their marginal alteration frequencies in a cohort of patients. However, the highly diverse alteration landscape characterizing malignant tumors is often poorly explained solely by the highly frequently altered genes (Stratton et al., 2009). A more sensitive and robust identification of driver events consists in analyzing the joint effect of multiple alterations performing the same functional role in tumor progression, commonly referred to as pathways. Once one of the members of a certain pathway is altered, tumors gain a significant selective advantage, which is often not further increased by the alteration of additional pathway members. The clones with the first occurring alteration likely become dominant, rendering all alterations in the same pathway to display a mutual exclusivity pattern across samples. Therefore, identifying groups of mutually exclusive alterations is a promising approach to identifying cancer drivers.
Several computational methods to identify mutually exclusive pathways either de novo (Vandin et al., 2012; Leiserson et al., 2013; Jerby-Arnon et al., 2014; Szczurek and Beerenwinkel, 2014; Constantinescu et al., 2015; Kim et al., 2015; Wu et al., 2015) or based on known biological interaction networks (Ciriello et al., 2012; Babur et al., 2015) have recently been developed. Multidendrix (Leiserson et al., 2013) employs integer linear programming to find multiple driver pathways, improving on Dendrix (Vandin et al., 2012), which identifies a single pathway with both high coverage and high exclusivity. CoMEt (Wu et al., 2015) further improves the methodology of Multidendrix by proposing a generalization of Fisher's exact test for evaluating mutual exclusivity. Muex (Szczurek and Beerenwinkel, 2014) is a statistical model in which members of mutually exclusive groups are required to have similar alteration frequencies, MEMCover (Kim et al., 2015) detects dysregulated mutually exclusive subnetworks across different cancer types, and Daisy (Jerby-Arnon et al., 2014) employs a data-driven approach for identifying synthetic lethality interactions in cancer, which lead to mutual exclusivity. Finally, TiMEx (Constantinescu et al., 2015) is a generative probabilistic graphical model, which quantifies exactly the degree of mutual exclusivity for each identified pathway. Unlike the other approaches, TiMEx explicitly models tumorigenesis as a dynamic process that occurs over long periods of time, accounting for the interplay between the waiting times to alteration of each gene and the observation time.
The progression of cancer can happen along various alternative evolutionary paths (Beerenwinkel et al., 2015; Diaz-Uriarte, 2015). In this context, Fearon and Vogelstein (1990) were the first to show that not all progression paths are equally probable. Based on genetic and clinical data, their conceptual model proposed the existence of a linear order among groups of mutations in colorectal cancer. Oncogenetic trees (Desper et al., 1999) generalized the idea of a single progression path, by allowing a diverging temporal ordering of events, under the assumption that each event depends on a single parent. Bayesian networks for cancer progression (Hjelm et al., 2006; Beerenwinkel et al., 2007; Beerenwinkel and Sullivant, 2009; Gerstung et al., 2009; Sakoparnig and Beerenwinkel, 2012) further relaxed the assumption of not allowing different progression paths to converge. For example, conjunctive Bayesian networks (CBNs) (Beerenwinkel and Sullivant, 2009; Gerstung et al., 2009) are generative probabilistic graphical models of tumor progression, defined by a partial order on a set of genetic events, in which the occurrence of each event can depend on more than one parent. Other approaches include RESIC (Attolini et al., 2010), which explicitly considers the evolutionary dynamics of mutation fixation; Progression Networks (Farahani and Lagergren, 2013), which employ mixed integer linear programming; and CAPRESE (Loohuis et al., 2014) and CAPRI (Ramazzotti et al., 2015), which use a framework of probability causation.
Virtually all cancer progression models are designed to infer tumorigenesis from cross-sectional data, that is, single-time snapshots from multiple patients. However, because of the very high interpatient heterogeneity (Vogelstein et al., 2013), carcinogenesis often follows different progression paths in different individuals. Even though the selective pressure in cancer is known to be stronger on pathways than on single alterations (Gerstung et al., 2011; Vogelstein et al., 2013), only few models describe tumorigenesis at the level of pathways (Gerstung et al., 2011; Cheng et al., 2012; Raphael and Vandin, 2014). Commonly, in a first step, single alterations are mapped to predefined pathways, and in a second step, progression algorithms are used to infer the order constraints among pathways (Gerstung et al., 2011; Cheng et al., 2012). Besides the inherent disadvantages of using predefined pathways, such as lack of specificity and large pathway size, these approaches are assuming that the inferences of either driver pathways or order constraints are independent of each other. However, Raphael and Vandin (2014) have devised a joint integer linear programming model of progression among mutually exclusive pathways, on the basis of which they have formally shown that the a priori knowledge of either pathways or progression is not sufficient for inferring the correct joint solution. Their model assumes that cancer progresses via a single linear path, which is a very restricted representation of tumorigenesis, particularly in light of previous findings (Gerstung et al., 2011; Cheng et al., 2012).
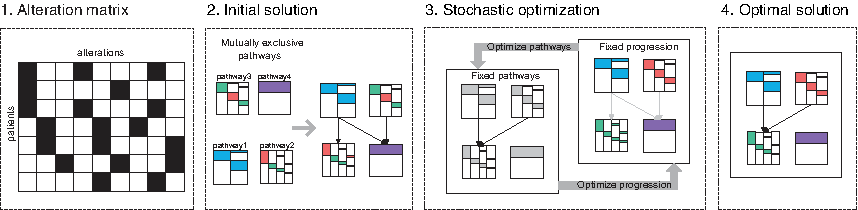
In this study, we introduce pathTiMEx, a generative probabilistic graphical model of cancer progression at the level of driver pathways (Fig. 1). pathTiMEx directly generalizes TiMEx (Constantinescu et al., 2015), a waiting time model for mutually exclusive cancer alterations, and CBN (Gerstung et al., 2009), a waiting time model for cancer progression (Fig. 2). We assume that in tumor development, alterations can either occur independently or depend on each other by being part of the same pathway or by following particular progression paths. By inferring these two types of potential dependencies simultaneously, pathTiMEx jointly identifies driver events and the evolutionary order constraints among them. In our approach, the structure among pathways is modeled as a directed acyclic graph (DAG), hence the model in Raphael and Vandin (2014) is a special case of pathTiMEx, corresponding to the situation where the structure is restricted to be a linear path.
Overview of pathTiMEx. In the first step, a cancer genomics dataset (consisting of, e.g., point mutations or copy number aberrations for a cohort of patients) is preprocessed in the form of a binary alteration matrix, with rows representing patients and columns representing alterations. A black square encodes the presence of an alteration, and a white square indicates its absence. In the second step, on the basis of the binary matrix, mutually exclusive groups of alterations (pathways) are inferred with TiMEx (Constantinescu et al., 2015), and the progression among pathways is inferred with CBN (Gerstung et al., 2009). In the third step, the progression among pathways is used as the initial solution to a stochastic optimization scheme, which consists of two parts. First, given the fixed optimal progression among pathways, the assignment of genes to pathways is optimized through a Markov Chain Monte Carlo approach (Madigan et al., 1995). Second, given the fixed optimal assignment of genes to pathways, progression among pathways is optimized through simulated annealing (Kirkpatrick et al., 1983). The joint optimization is repeated until both the assignment of genes to pathways and the progression among pathways converge to the joint optimal solution.
pathTiMEx as a probabilistic graphical model. Nodes represent random variables, while directed edges represent their dependencies. In this example, there are \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n = 8$$
\end{document} genes partitioned into \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$k = 3$$
\end{document} pathways as \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$G = \{ 1 , \ldots , 8 \} $$
\end{document} = \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ 1 , 2 \} \cup \{ 3 , 4 , 5 , 6 \} \cup \{ 7 , 8 \} $$
\end{document}. Different colors (blue, red, and green) correspond to different pathways. The hidden variables, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$T = ( {T_1}, \ldots , {T_8} )$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$U = ( {U_1},{U_2},{U_3} )$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${T_{\rm obs}}$$
\end{document}, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$X = ( {X_1}, \ldots , {X_8} )$$
\end{document}, are depicted as empty circles, while the observed ones, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$Y = ( {Y_1}, \ldots {Y_8} )$$
\end{document}, are depicted as filled circles. Variables Tg are the waiting times to alteration of the genes \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$g \in G$$
\end{document}, Up are the waiting times to alteration of the pathways \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \in { \cal P}$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${T_{{ \rm{obs}}}}$$
\end{document} is the observation time, Xg are the true mutational statuses of the genes, and Yg are the observed ones.
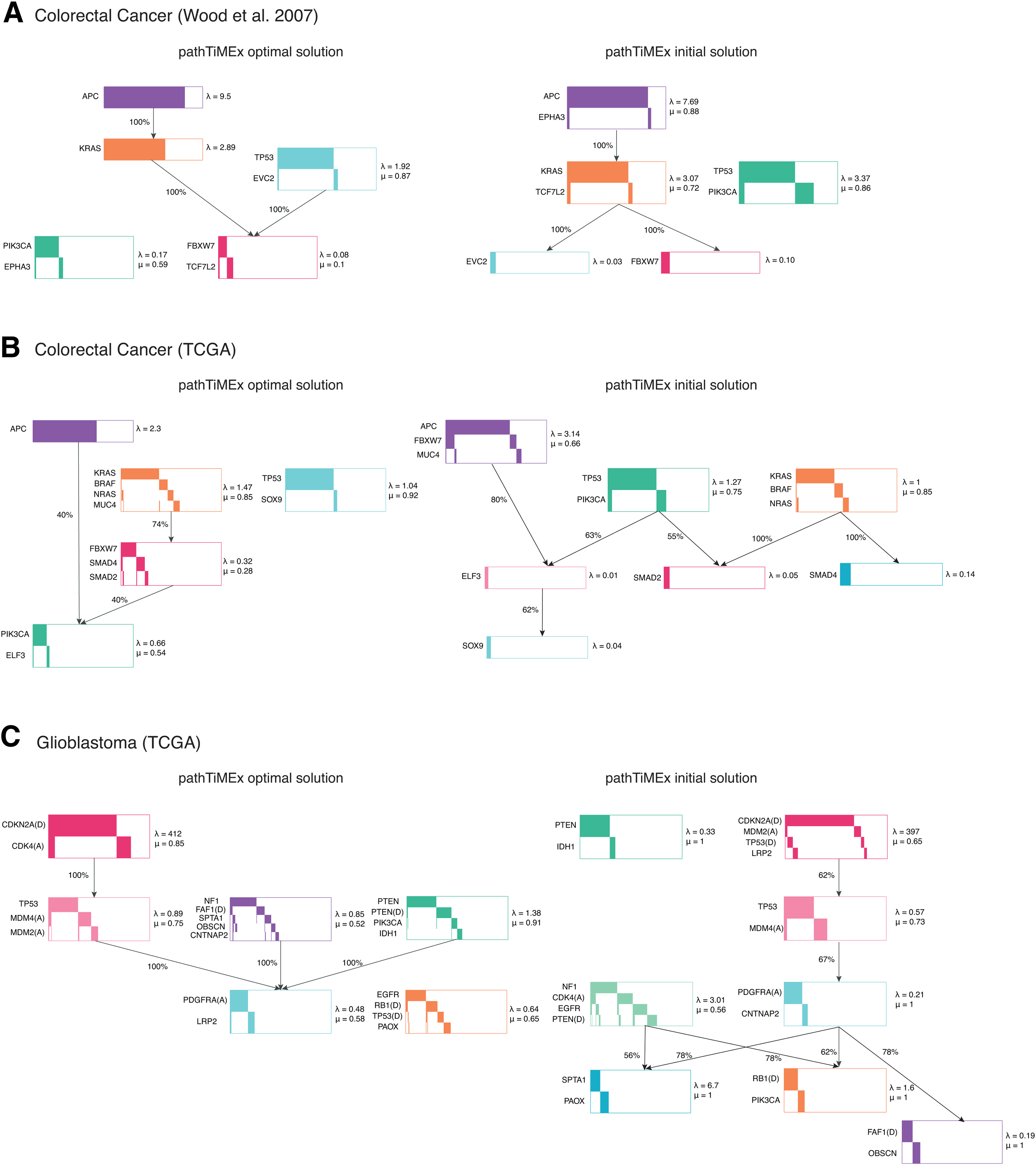
On real cancer data, pathTiMEx recapitulates previous knowledge on tumorigenesis such as the temporal order among pathways which include APC, KRAS, and TP53 in colorectal cancer, while also proposing new biological hypotheses, such as the existence of a single early causal event consisting of the amplification of CDK4 and the deletion of CDKN2A in glioblastoma. pathTiMEx is available as an R package at https://github.com/cbg-ethz/pathTiMEx.
2. Methods
2.1. Genes and pathways
Let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$G = \{ 1 , \ldots , n \} $$
\end{document} be a set of n genes. A partition of G is a collection, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document}, of subsets of G such that (1) \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document} does not contain the empty set, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\emptyset \;\notin\; { \cal P}$$
\end{document}; (2) the union of all elements of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document} is equal to G, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\bigcup \nolimits_{P \in { \cal P}} P = G$$
\end{document}; and (3) the elements of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document} are pairwise disjoint, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \cap Q = \emptyset$$
\end{document}, for all \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \neq Q \in { \cal P}$$
\end{document}. The cardinality of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document} is denoted by k. The elements of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document} are sets of genes and will be referred to as pathways. Every gene \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$g \in G$$
\end{document} belongs to exactly one pathway denoted by \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_g} \in { \cal P}$$
\end{document}.
The genotype of a tumor is defined by the set of genes that are altered (alternatively referred to as mutated) in the tumor. We represent genotypes as binary vectors \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$x = ( {x_1}, \ldots , {x_n} )$$
\end{document} of length n, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${x_i} = 1$$
\end{document} indicates the presence of a mutation in gene i and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${x_i} = 0$$
\end{document} indicates its absence. Equivalently, a genotype x can be defined as the subset of mutated genes \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${G_x} = \{ g \in G \vert {x_g} = 1 \} $$
\end{document}. For a partition \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document}, we have \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${G_x} = \bigcup \nolimits_{P \in { \cal P}} \left( {P \cap {G_x}} \right)$$
\end{document}, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \cap {G_x}$$
\end{document} is the set of mutated genes in pathway P.
To model tumor progression, we consider a partial order among pathways of a given partition \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document}. Intuitively, we require pathway P to be altered before pathway Q whenever P is smaller than Q in the partial order. Formally, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( { \cal P}, \prec )$$
\end{document} is a partially ordered set, or poset, if \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\prec$$
\end{document} is a reflexive, antisymmetric, and transitive relation on \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document}. For any pathway Q, we define the set of parents of Q as \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\rm pa} ( Q ) = \{ P \in { \cal P} \vert P \prec Q$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$ \nexists P^{\prime} \in { \cal P}, P^{\prime} \ne P , Q \ { \rm{with }} \ P \prec P^{\prime} \prec Q \} $$
\end{document}.
2.2. Waiting times
We describe the time to occurrence of an alteration of gene \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$g \in G$$
\end{document} by the random variable Tg, and similarly the waiting time to alteration of pathway \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \in { \cal P}$$
\end{document} by UP. A pathway is assumed to be altered as soon as one of its member genes is altered. Hence, the pathway alteration time is the minimum of the alteration times of its genes,
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{U_P} = \mathop { \min } \limits_{g \in P} { \kern 1pt} {T_g}. \tag{1}
\end{align*}
\end{document}
The poset structure among pathways implies that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${U_P} \le {U_Q}$$
\end{document} whenever \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \prec Q$$
\end{document}. This constraint is modeled by
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{T_g} \sim \mathop { \max } \limits_{Q \in {\rm pa} ( {P_g} ) } {U_Q} + {\rm Exp} ( { \lambda _g} ), \tag{2}
\end{align*}
\end{document}
where Exp denotes the exponential distribution.
Thus, the total waiting time to alteration for gene g, in pathway Pg, is the time it takes for all predecessor pathways to mutate plus the individual waiting time contribution of g, which is drawn from an exponential distribution with rate \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _g}$$
\end{document}. The gene waiting times, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$T = ( {T_1}, \ldots , {T_n} )$$
\end{document}, and the pathway waiting times, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$U = ( {U_1}, \ldots , {U_k} )$$
\end{document}, are well defined by Equations (1) and (2) if pathways are visited according to a topological sort of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( { \cal P}, \prec )$$
\end{document}.
Both T and U are hidden random variables because we do not observe the relative time at which genes or pathways are altered, relative to cancer initiation. Instead, we have cross-sectional data reporting the set of mutations in a cohort of patients, accumulated up to the time of diagnosis, which is usually different for each tumor. To represent this process explicitly, we model the observation time as an exponentially distributed random variable:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{T_{\rm obs}} \sim {\rm Exp} ( { \lambda _{\rm obs}} ). \tag{3}
\end{align*}
\end{document}
Specifically, the observation time is the time at which the tumor biopsy is taken relative to tumor initiation. Because the onset of tumorigenesis is generally unknown, the observation time is also an unobserved random variable.
2.3. Mutual exclusivity
In general, a gene g can only be mutated in a given genotype if the biopsy has been taken after the mutation occurred, that is, if \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${T_g} < {T_{\rm obs}}$$
\end{document}. However, due to mutual exclusivity, the mutation may not be present in the genotype if another gene from the same pathway was mutated before. The underlying rationale is that genes in the same pathway contribute to the same important function for tumor development, such that the alteration of a single pathway member is sufficient for cancer to progress. In our framework, the alteration to be observed will be the one with the shortest waiting time among all genes in a pathway. To formalize this dependency of genotypes on waiting times, let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$X = ( {X_1}, \ldots , {X_n} )$$
\end{document} be the vector of binary random variables describing the genotype \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$x = ( {x_1}, \ldots , {x_n} )$$
\end{document}.
We denote by \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _P}$$
\end{document} the probability that pathway P is perfectly mutually exclusive (no two genes in P share mutations in the same tumor) or, equivalently, the degree of mutual exclusivity of pathway P. With probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _P}$$
\end{document}, alterations in additional pathway members do not fixate before observation time. Gene g is mutated \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( {X_g} = 1 )$$
\end{document} with probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _{{P_g}}}$$
\end{document} if pathway P is mutually exclusive and g was its first member to mutate (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${U_{{P_g}}} = {T_g}$$
\end{document}) and with probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 1 - { \mu _{{P_g}}} )$$
\end{document} if P is not mutually exclusive and g just happened to mutate first. Thus, for each \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$g \in G$$
\end{document}, we draw Xg from the following Bernoulli distribution:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\left( {{X_g} \vert T , U , {T_{\rm obs}}} \right) \sim {\rm Bern} \left( {{ \mathbb{I}_{{T_g} < {T_{\rm obs}}}} ( { \mu _{{P_g}}}{ \mathbb{I}_{{U_{{P_g}}} = {T_g}}} + 1 - { \mu _{{P_g}}} ) } \right), \tag{4}
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mathbb{I}$$
\end{document} is the indicator variable, that is, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mathbb{I}_A} = 1$$
\end{document} if statement A is true and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mathbb{I}_A} = 0$$
\end{document} otherwise.
In real datasets, we may not observe the true genotype X because of false positive or false negative mutation calls. We model noisy observations by introducing the binary random variables \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$Y = ( {Y_1}, \ldots , {Y_n} )$$
\end{document}, generated from X by independently flipping each bit with probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document},
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\left( {{Y_g} \vert {X_g}} \right) \sim {\rm Bern} \left( {{{ ( 1 - \varepsilon ) }^{{X_g}}}{ \kern 1pt} { \varepsilon ^{1 - {X_g}}}} \right), \tag{5}
\end{align*}
\end{document}
for all genes \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$g \in G$$
\end{document}.
pathTiMEx is the generative probabilistic model for the hidden random variables T, U, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${T_{\rm obs}},$$
\end{document} and X, and the observed random variable Y, defined by Equations (1–5), with parameters \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\theta = ( { \cal P}$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\prec$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _1}, \ldots , { \lambda _n}$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _{\rm obs}}$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _1}, \ldots , { \mu _{{k}}}$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document}). The statistical dependencies among the random variables of pathTiMEx, represented as a probabilistic graphical model, are displayed in Figure 2.
The model can be regarded as a generalization of both CBNs (Beerenwinkel et al., 2007; Beerenwinkel and Sullivant, 2009; Gerstung et al., 2009), which have been used to model partial order constraints among a given set of predefined pathways (Gerstung et al., 2011), and TiMEx, a waiting time model for mutually exclusive mutations (Constantinescu et al., 2015).
2.4. Further model specifications
Similarly to related models (Beerenwinkel et al., 2007; Constantinescu et al., 2015), not all parameters of pathTiMEx can be identified from cross-sectional datasets which lack information about the absolute timescale of cancer progression. However, for fixed \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _{\rm obs}}$$
\end{document}, the model becomes identifiable. Hence, without loss of generality, we assume \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _{\rm obs}} = 1$$
\end{document} from now on.
pathTiMEx accounts for imperfect mutual exclusivity through the parameter \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _P}$$
\end{document}, and for erroneous observations through the parameter \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document}. To simplify the model, we lump the biological and technical noise sources together in the parameter \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document}. Specifically, we set \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _P} = 1$$
\end{document} for all \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \in { \cal P}$$
\end{document}. With this assumption, Equation (4) simplifies to
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{X_g} = { \mathbb{I}_{{T_g} < {T_{\rm obs}}}}{ \mathbb{I}_{{U_{{P_g}}} = {T_g}}}. \tag{6}
\end{align*}
\end{document}
2.5. Likelihood
For the pathTiMEx model \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\theta$$
\end{document}, given that obs is the observation event and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$t , u \ \ { \rm{and}} \ \ {t_{{ \rm{obs}}}}$$
\end{document} are instantiations of the random variables \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$T , U \ \ { \rm{and}} \ \ {T_{{ \rm{obs}}}}$$
\end{document}, the joint probability density of the waiting times is
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\begin{split}{f_{T , U , {T_{{ \rm{obs}}}}}} \left( {t , u ,
{t_{{ \rm{obs}}}} \vert \theta } \right) & = \prod \limits_{g \in
G} {f_{{T_g}}} \left( {{t_g} \vert {U_i} = {u_i}, \forall i \in {
\rm{pa}} ( {P_g} ), \theta } \right) \times \prod \limits_{P \in {
\cal P}} {f_{{U_P}}} \left( {{u_P} \vert {T_j} = {t_j}, \forall j
\in P , \theta } \right) {f_{{T_{{ \rm{obs}}}}}} \left( {{t_{{
\rm{obs}}}} \vert \theta } \right) \\ & = \prod \limits_{g \in \{
G \cup obs \} } { \lambda _g}{e^{ - { \lambda _g} \left( {{t_g} -
\mathop { \max } \limits_{i \in { \rm{pa}} ( {P_g} ) } \mathop {
\min } \limits_{j \in i} {t_j}} \right) }} \times {1_{{t_g} >
\mathop { \max } \limits_{i \in { \rm{pa}} ( {P_g} ) } \mathop {
\min } \limits_{j:{P_j} = i} {t_j}}}.\end{split}
\tag{7}\end{align*}
\end{document}
Conditioned on the observation time and on the waiting times of the pathways, the true mutational statuses of genes in separate pathways are independent. Their conditional probability distribution is
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
P \left( {X \vert T , U , {T_{{ \rm{obs}}}}, \theta } \right) = \prod \limits_{g \in G} P \left( {{X_g} \vert {T_g},{U_{{P_g}}},{T_{{ \rm{obs}}}}, \theta } \right) = \prod \limits_{P \in { \cal P}} P \left( {{G_x} \cap P \vert {T_k}, \forall k \in P , {T_{{ \rm{obs}}}}, \theta } \right). \tag{8}
\end{align*}
\end{document}
In the absence of noise, in a given pathway P, a true genotype contains no mutations if the observation time is shorter than the waiting times of all pathway members. Since we assume \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _P} = 1$$
\end{document}, a single mutation (of gene j) is present in the set \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${G_x} \cap P$$
\end{document} if and only if P is perfectly mutually exclusive. The presence of additional mutations in P represents a deviation from mutual exclusivity and, in the absence of noise, has probability 0:
The likelihood of the true genotype X, is the marginal probability
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
P \left( {X \vert \theta } \right) = \mathop \int \nolimits_{ \mathbb{R}_{ \ge 0}^{n + k + 1}} \cdots \int {f_{T , U , {T_{{ \rm{obs}}}}}} \left( {t , u , {t_{{ \rm{obs}}}} \vert \theta } \right) P \left( {X \vert T , U , {T_{{ \rm{obs}}}}, \theta } \right) { \kern 1pt} { \rm{d}}t \;{ \rm{d}}u \;{ \rm{d}}{t_{{ \rm{obs}}}}, \tag{10}
\end{align*}
\end{document}
which can be decomposed into a sum over all linear extensions of the poset \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( { \left( {{ \cal P} \cup { \rm{obs}}} \right), \prec } \right)$$
\end{document} (proof in Supplementary Section S1.1). Conditioned on the true genotype X, the likelihood of the observed genotype Y is
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
P \left( {Y \vert X , \theta } \right) = \prod \limits_{g \in G} P \left( {{Y_g} \vert {X_g}, \theta } \right) = { \varepsilon ^{{ \rm{d}} \left( {Y - X} \right) }}{ \left( {1 - \varepsilon } \right) ^{n - { \rm{d}} \left( {Y - X} \right) }}, \tag{11}
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \rm{d}} \left( {X , Y} \right) = \mathop \sum \nolimits_{g \in G} \vert {Y_g} - {X_g} \vert$$
\end{document} is the Hamming distance between the true genotype X, and the observed genotype Y. If we denote by \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$J \left( { \cal P} \right)$$
\end{document} the set of all true genotypes which could have been generated by the poset \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( { \left( {{ \cal P} \cup { \rm{obs}}} \right), \prec } \right)$$
\end{document}, it follows that the likelihood of the observed genotype Y is
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
P \left( {Y \vert \theta } \right) = \mathop \sum \limits_{X \in J \left( { \cal P} \right) } P \left( {Y \vert X , \theta } \right) P \left( {X \vert \theta } \right). \tag{12}
\end{align*}
\end{document}
The log likelihood of a dataset of N independent samples, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{Y}} = \left( {{Y^{ ( 1 ) }}, \ldots , {Y^{ ( N ) }}} \right)$$
\end{document}, where each \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${Y^{ ( i ) }}$$
\end{document} is an observed genotype, is
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
l \left( \theta \right) = \mathop \sum \limits_{i = 1}^N \log P \left( {{Y^{ ( i ) }} \vert \theta } \right). \tag{13}
\end{align*}
\end{document}
2.6. Inference
The inference scheme of pathTiMEx aims to maximize the log likelihood in Equation (13). The initial solution of our inference procedure is generated by first running TiMEx (Constantinescu et al., 2015) with default parameters iteratively on the input binary dataset. Each time, the largest and most significant pathway is retained and TiMEx is run again on the dataset from which the members of the identified pathway were excluded. This procedure is repeated until no more significantly mutually exclusive pathways are found. TiMEx identifies mutually exclusive groups as maximal cliques and estimates \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _g}$$
\end{document}, the exponential waiting time rates of each gene, as well as the mutual exclusivity intensities of each pathway, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _P}$$
\end{document}. These parameters are approximated with their maximum likelihood estimates, computed numerically. CBN inference (Gerstung et al., 2009) is then run on the binarized matrix obtained by encoding a pathway as altered whenever at least one of its gene members is altered. CBN estimates \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _P}$$
\end{document}, the exponential waiting time rates of each pathway, as well as the error probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document}, through a nested expectation-maximization (EM) algorithm (Dempster et al., 1977). Specifically, conditioned on an optimal fixed value of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _p}$$
\end{document} are optimized and, conditioned on the optimal fixed values of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \lambda _p}$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document} is optimized. Finally, the progression among pathways is inferred through simulated annealing (Kirkpatrick et al., 1983).
Starting from the initial solution, the joint optimization of pathways and structure follows a framework similar to an EM algorithm: conditioned on the optimal assignment of genes to pathway \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \cal P}$$
\end{document}, the poset \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( {{ \cal P}, \prec } \right)$$
\end{document} is optimized and, conditioned on the optimized poset, a new optimal assignment is computed. The procedure is repeated until both the assignment and the structure converge. Specifically, in iteration \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$i \ge 2$$
\end{document}, given an optimal fixed structure \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( {{{ \widehat { \cal P}}_{i - 1}},{{ \widehat { \prec} }_{i - 1}}} \right)$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${{ \cal P}_i}$$
\end{document} is optimized through a Markov Chain Monte Carlo (MCMC) scheme (Madigan et al., 1995). This amounts to minimizing the number of contradictions due to both mutual exclusivity and progression, Ei, which represents the number of ones that would need to be changed to zeroes so that the data \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{Y}}$$
\end{document} is consistent with both the assignment of genes to pathways Pi, and the poset \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( {{{ \widehat { \cal P}}_{i - 1}},{{ \widehat { \prec} }_{i - 1}}} \right)$$
\end{document}. For fixed parameters \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( {{ \cal P}, \prec } \right)$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda$$
\end{document}, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mu = 1$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${E_i} = \mathop \sum \nolimits_{j = 1}^N { \rm{d}} \left( {{X^j},{Y^j}} \right)$$
\end{document}, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{X}} = \left( {{X^{ ( 1 ) }}, \ldots , {X^{ ( n ) }}} \right)$$
\end{document} is the set of true genotypes. Therefore, minimizing Ei is equivalent to maximizing the likelihood in Equation (13) as a function of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \widehat E_i} = Nn \;{ \widehat \varepsilon _i}$$
\end{document} (proof in Supplementary Section S1.2). A detailed explanation of the MCMC scheme, including its computational complexity, is given in Supplementary Section S1.2.
Given the optimal assignment \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\widehat {{P_i}}$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( {{{ \cal P}_i},{ \prec _i}} \right)$$
\end{document} is optimized through simulated annealing by locally maximizing the likelihood function in Equation (13), given the optimal set of parameters \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\widehat {{ \lambda _p}}$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\widehat \varepsilon$$
\end{document}, found through the nested EM procedure. Starting from a poset P, a new poset \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P^{\prime}$$
\end{document} is proposed and it is accepted either if it increases the likelihood or, otherwise, with probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \rm{exp}} \left( { - \left[ {l \left( { \widehat {{ \lambda _p}}, \widehat \varepsilon , P} \right) - l \left( { \widehat {{ \lambda _p}}, \widehat \varepsilon , P^{\prime} } \right) } \right] / C} \right)$$
\end{document}. The temperature C reduces the risk of remaining in local optima.
3. Results
3.1. Simulations
We compared the performance of the iterative and the classic TiMEx procedures using simulated data for various noise levels \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document} and number of samples N. Specifically, we considered two independent mutually exclusive groups consisting of two genes each, for which the waiting time rates \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda$$
\end{document} were uniformly sampled between \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.1$$
\end{document} and 2. In this setting, the genes cover a wide range of alteration frequencies, from less than \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$1 \%$$
\end{document} to more than \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$60 \%$$
\end{document} (Constantinescu et al., 2015). In the absence of noise, all pathways were perfectly mutually exclusive, that is, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mu = 1$$
\end{document}. With error probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon \in \{ 0 , 0.05 , 0.1 , 0.15 \} $$
\end{document}, each entry in the binary alteration matrix consisting of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N \in \{ 100 , 400 , 1000 \} $$
\end{document} patients was flipped either from zero to one or otherwise. We generated 100 datasets corresponding to each of the above configurations and inferred mutually exclusive groups with either the iterative or the classic TiMEx procedures. Iterative TiMEx clearly outperformed classic TiMEx for all tested sample sizes and noise levels (Supplementary Fig. S1).
Next, we designed a simulation experiment assessing the performance and stability of pathTiMEx for the noise levels \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document} and sample sizes N mentioned above. We simulated a progression model consisting of 12 genes assigned to 5 mutually exclusive pathways. The assignment of genes to pathways was random, with the sole restriction that each pathway contained at least one gene. 87% of the 100 generated datasets included at least one pathway with a single member and 78% of them included at least one pathway with at least four members. The order constraints among pathways were generated as a DAG with a number of edges uniformly sampled as to satisfy an edge density (number of existing edges divided by number of all possible edges) of between \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.4$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.8$$
\end{document}. In consequence, each poset had at least one unconnected pathway, and the majority of pathways were connected to either one or two other pathways. Our simulation scenarios resemble real cancer datasets in terms of number of pathways, alteration frequencies, noise levels, and the presence of pathways either consisting of single genes or unconnected to other pathways (Gerstung et al., 2011; Raphael and Vandin, 2014). On all simulated datasets, we jointly inferred mutually exclusive pathways and the order constraints among them with pathTiMEx. We compared our final results with the initial solution of pathTiMEx, that is, iteratively identifying mutually exclusive groups with TiMEx (Constantinescu et al., 2015), followed by optimizing the order dependencies among the groups a single time with CBN (Gerstung et al., 2009). As an implementation of the model proposed in Raphael and Vandin (2014) is not publicly available, we were not able to compare our results with their approach on simulated data.
The convergence rate of pathTiMEx increases with increasing sample size and decreasing noise levels (Supplementary Table S1). For \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon = 0$$
\end{document}, the algorithm converged in all cases for all sample sizes, while for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon = 0.1$$
\end{document}, it converged in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$51 \%$$
\end{document} of the runs for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 100$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$97 \%$$
\end{document} for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 400$$
\end{document}, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$98 \%$$
\end{document} for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 1000$$
\end{document}. Similarly, up to random fluctuations, the number of iterations required for convergence decreases with increasing sample size and decreasing noise levels. When \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 400$$
\end{document}, pathTiMEx converges in an average of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$2.6$$
\end{document} iterations for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon = 0$$
\end{document}, compared with an average of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$12.5$$
\end{document} iterations when \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon = 0.15$$
\end{document}. The runtime per iteration increases with increasing sample size and remains largely unaffected by noise, with the exception of the largest sample size. The decrease in runtime per iteration with increasing noise level when \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 1000$$
\end{document} can be explained by a slight, but noticeable, increase in the false-negative rate.
pathTiMEx identified the correct assignment of genes to pathways in the largest percentage of cases (Supplementary Fig. S2A). The average Rand index (Rand, 1971) between the true pathways and the estimated ones ranged from \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.95$$
\end{document} for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon = 0$$
\end{document} to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.84$$
\end{document} for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon = 0.15$$
\end{document} in the case of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 100$$
\end{document} (with corresponding Jaccard indices [Levandowsky and Winter, 1971] of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.75$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.44$$
\end{document}) and from \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.99$$
\end{document} to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.92$$
\end{document} when \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 1000$$
\end{document} (with corresponding Jaccard indices of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.94$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.68$$
\end{document}). pathTiMEx outperformed the initial solution in almost all situations, with the exception of large sample sizes and a large noise level. In these cases, as previously mentioned, the joint effect of large sample size and low progression signal led to an increase in both false positive and false negative rates. For all sample sizes and noise levels, among the cases in which the genes were correctly assigned to pathways (clustering similarity indices of 1), pathTiMEx further identified the correct progression structure in the large majority of cases (Supplementary Fig. S2B). Structure similarity increases with increasing sample size and decreasing noise levels.
3.2. Cancer data
We used pathTiMEx to jointly infer mutually exclusive driver pathways and order constraints among them in three publicly available cancer datasets, namely a small colorectal cancer dataset (Wood et al., 2007), a large colorectal cancer dataset from TCGA (Cerami et al., 2012), and a large glioblastoma dataset from TCGA (Cerami et al., 2012). We compared our results with the basic approach of decoupling the identification of mutually exclusive groups and the inference of their progression, which is the initial solution to our stochastic joint optimization scheme. Additionally, as the approach developed in Raphael and Vandin (2014) is a special case of pathTiMEx, we facilitated a direct comparison between the two tools by optimizing the assignment of genes to pathways under a fixed linear progression. Under these constraints, pathTiMEx is much faster than the algorithm in Raphael and Vandin (2014), as solely optimizing mutual exclusivity for a fixed progression involves one single iteration of the MCMC chain.
For the three datasets, we assessed the stability of the optimal pathTiMEx solutions across 100 runs, and in each run, the joint optimization procedure was iterated at most 100 times (Supplementary Table S2). For each identified order dependency among pathways, we computed its frequency across all runs. As the initial pathways were fixed in all cases, the edge weights in the initial solutions only evaluate the stability of the order constraints and are usually higher. Additionally, the estimation of the rate \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda$$
\end{document} and the error probability \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\varepsilon$$
\end{document} was highly stable (less than \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.01 \%$$
\end{document} variance across runs). According to our model, high rates of evolution (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda$$
\end{document}) indicate events that happened soon after their parents have occurred, while low rates indicate events that happened later.
3.3. Colorectal cancer
The colorectal cancer dataset published in Wood et al. (2007) consists of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n = 8$$
\end{document} genes mutated with frequency above \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$5 \%$$
\end{document} in 95 samples. This dataset has been previously used in Gerstung et al. (2011) and Raphael and Vandin (2014) for analyzing cancer progression among pathways. The optimal structure inferred by pathTiMEx (Fig. 3A) reaffirmed the current knowledge on tumor progression in colorectal cancer: APC is an early event (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 9.5$$
\end{document}), followed by KRAS\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( \lambda = 2.89 )$$
\end{document} and TP53\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( \lambda = 1.92 )$$
\end{document}. pathTiMEx immediately converged in all 100 runs to the same joint solution (Supplementary Table S2), with an estimated \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\widehat \varepsilon$$
\end{document} of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.13$$
\end{document}.
Results of pathTiMEx on cancer datasets. The optimal solution inferred by pathTiMEx is shown on the left-hand side for three cancer datasets. The basic approach (right-hand side) assumes that pathways and progression are decoupled and it represents the initial solution of our stochastic joint optimization scheme. Each pathway is further characterized by the estimates of its waiting time rate \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda$$
\end{document}, and its mutual exclusivity intensity \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mu$$
\end{document}. The weight on each edge shows how likely it is to infer a directed dependency between the same two groups, across 100 runs. The percentage is computed relative to all the cases in which the algorithm converged in less than 100 iterations (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$100 / 100$$
\end{document} runs for the colorectal cancer dataset in Wood et al. [2007], \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$91 / 100$$
\end{document} runs for the TCGA colorectal cancer dataset, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$26 / 100$$
\end{document} runs for glioblastoma). In the glioblastoma dataset, (D) indicates the deletion of a gene, while (A) indicates its amplification.
None of the five identified mutually exclusive pathways were also identified by the basic approach, that is, they were not part of the initial solution, which means that mutual exclusivity and progression are not independent. For example, the basic approach estimated that the group TP53 and PIK3CA is independent and it occurs earlier in progression than KRAS and TCF7L2. The same temporal order was inferred in Raphael and Vandin (2014) and also by pathTiMEx if assuming a linear structure (Fig. 4A). On the contrary, pathTiMEx identified TP53 and EVC2 (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 1.92$$
\end{document}) as a later event than KRAS, which is consistent with the current knowledge on colorectal tumorigenesis (Fearon and Vogelstein, 1990; Fearon, 2011). Additionally, by enforcing a linear order among pathways, our model obtained a better score (the number of mismatches due to violation of both mutual exclusivity and progression constraints) than Raphael and Vandin's approach (Fig. 4A). The late events found by pathTiMEx were PIK3CA and EPHA3 (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 0.17$$
\end{document}), together with FBXW7 and TCF7L2 (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 0.08$$
\end{document}). These findings, similar to the ones obtained if assuming a linear progression, confirm previous observations in Gerstung et al. (2011).
Comparison between pathTiMEx and the approach of Raphael and Vandin (2014) on (A) the colorectal cancer dataset in Wood et al. (2007) and (B) the colorectal cancer dataset from TCGA. To facilitate a direct comparison between the two models, we optimized the assignment of genes to pathways with pathTiMEx, assuming a fixed linear progression with an unspecified number of stages. We computed scores for the two models as the number of mismatches due to violation of both mutual exclusivity and progression constraints. pathTiMEx estimates four progression stages on both colorectal cancer datasets and performs better than the model in Raphael and Vandin (2014) by identifying pathways with smaller numbers of mismatches. In (B), differences in data preprocessing led to the gene MUC4 only being present in pathTiMEx, and the genes FAM123B and TCFL72 only being present in the model from Raphael and Vandin (2014).
The gene-level model of tumor progression proposed in Gerstung et al. (2011) also identified APC as an early event, followed by a pathway, including KRAS and TP53, which was unconnected to other pathways. However, their pathway-level model of cancer progression employed large predefined pathways and did not attempt to identify pathways de novo.
3.4. Colorectal cancer (TCGA)
For \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 258$$
\end{document} colorectal cancer samples, we analyzed point mutation data for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n = 473$$
\end{document} genes, either significantly recurrently mutated as identified by MutSigCV (Lawrence et al., 2013) or part of copy number aberrations as identified by GISTIC 2.0 (Mermel et al., 2011). In addition to the three mutually exclusive groups found by iterative TiMEx (Constantinescu et al., 2015), we introduced in our analysis four more genes with known involvement in colorectal tumorigenesis (Raphael and Vandin, 2014): ELF3, SOX9, SMAD2, and SMAD4. pathTiMEx converged in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$91 \%$$
\end{document} of the runs, with \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\widehat \varepsilon = 0.16$$
\end{document} (Supplementary Table S2). The most likely progression structure (Fig. 3B) was identified in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$40 \%$$
\end{document} (37) of the 91 runs. The average Jaccard index (Levandowsky and Winter, 1971) between the pathways alternatively reported and the most likely pathways was \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.81$$
\end{document} (with a minimum of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.66$$
\end{document}). Across all runs, provided that the reported pathways were also the most likely ones, the dependencies among them were identical.
The optimal structure inferred by pathTiMEx (Fig. 3B) was in high concordance with our findings on the colorectal cancer dataset analyzed above (Fig. 3A) and also consistent with the current knowledge on colorectal tumorigenesis (Fearon and Vogelstein, 1990; Fearon, 2011). Specifically, APC (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 2.3$$
\end{document}) was the earliest event, followed by a mutually exclusive pathway, including KRAS (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 1.47$$
\end{document}), and later followed by TP53 and SOX9 (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 1.04$$
\end{document}). Interestingly, on both colorectal cancer datasets, even if APC was initially part of a mutually exclusive group, in the optimal solution, it is the single early starting event. This finding emphasizes the particular importance that APC has in the initiation of colorectal cancer (Fearon and Vogelstein, 1990). As previously observed, the basic approach identified TP53 as an early event, and only following the joint optimization of mutual exclusivity and progression, mutations in TP53 were reported as later events. Most of the genes part of the identified mutually exclusive groups are known interaction partners in colorectal cancer, such as the tumor suppressors SMAD2 and SMAD4 (Raphael and Vandin, 2014). Interestingly, pathTiMEx identified MUC4, a gene that is aberrantly expressed in colorectal adenocarcinomas, but with an unknown prognostic value (Shanmugam et al., 2010), as part of the same mutually exclusive group with three oncogenes in the Ras-Raf pathway, namely BRAF, NRAS, and KRAS (Raphael and Vandin, 2014).
In conclusion, the results of pathTiMEx are consistent between the two colorectal cancer datasets and more consistent with the literature than previous approaches. By imposing a linear order, our model estimated a progression of four stages and obtained a better score than the model proposed in Raphael and Vandin (2014) (Fig. 4B), namely fewer mismatches due to violation of both mutual exclusivity and progression constraints. Hence, pathTiMEx offers a better explanation of colorectal tumorigenesis than the basic initial solution and the models in Gerstung et al. (2011) and Raphael and Vandin (2014).
3.5. Glioblastoma (TCGA)
We analyzed the glioblastoma dataset discussed in Leiserson et al. (2013) and preprocessed as explained in Constantinescu et al. (2015), consisting of point mutations and copy number aberrations for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n = 486$$
\end{document} genes in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N = 261$$
\end{document} patients. In glioblastoma, tumor progression is poorly understood and more difficult to infer than in colorectal cancer (Cheng et al., 2012). Consequently, our algorithm converged slower than in the case of the two colorectal cancer datasets (Supplementary Table S2), and the error rate was \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\widehat \varepsilon = 0.2$$
\end{document}. pathTiMEx only reached a stable solution in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$26 \%$$
\end{document} of the runs in less than 100 iterations. In these cases, it always converged to the same pathways and dependencies among them.
The optimal solution of pathTiMEx (Fig. 3C) consisted of fewer pathways than the initial solution. Unlike colorectal cancer, all members of the optimal pathways were part of large mutually exclusive groups, which points to the large variability of tumor progression in glioblastoma. Following the joint optimization scheme, pathTiMEx identified as a very early event (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 412$$
\end{document}) the group that included the deletion of CDKN2A and amplification of CDK4A. These two genes are interaction partners and belong to the pathways CDC42 signaling events and Cyclin D-associated events in G1. This group is directly temporally related to a later event consisting of the copy number amplifications of MDM4 and MDM2, together with the point mutation of TP53 (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 0.89$$
\end{document}), which are all members of the p53 pathway (Brennan et al., 2013) and were previously identified as playing a role in tumor progression in glioblastoma (Cheng et al., 2012; Raphael and Vandin, 2014). The point mutation of TP53 and the amplification of MDM4 were initially reported as a mutually exclusive pair by the basic approach. Following the joint optimization scheme, the pair was merged with the amplification of MDM2, a member of the same pathway. Interestingly, the point mutation and deletion of PTEN, together with the point mutation of PIK3CA (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda = 1.38$$
\end{document}), which belong to the PI3K pathway, were identified as part of a mutually exclusive group that also included IDH1.
In conclusion, despite the high variability in the data, pathTiMEx offers new insights into tumorigenesis in glioblastoma by jointly optimizing mutually exclusive pathways and the order constraints among them.
4. Discussion
We have introduced pathTiMEx, a probabilistic model of tumor progression among mutually exclusive driver pathways, together with an efficient stochastic joint optimization scheme. pathTiMEx is a step forward from approaches that separately infer either mutually exclusive groups of alterations or cancer progression among single genes. The simultaneous identification of driver pathways and evolutionary order constraints among them may have important therapeutic implications, particularly by targeting members of early mutually exclusive pathways (Weinstein, 2002).
pathTiMEx is a direct generalization of both TiMEx (Constantinescu et al., 2015), a waiting time model for independent mutually exclusive pathways, and CBN (Gerstung et al., 2009), a waiting time model for cancer progression among single genes. pathTiMEx assumes that in tumor development, alterations can occur independently or depend on each other by being part of the same pathway or by following particular progression paths. By inferring these two types of potential dependencies simultaneously, pathTiMEx jointly addresses the two fundamental questions of identifying drivers and progression. pathTiMEx models the order constraints among pathways as a DAG, hence the only previous approach performing simultaneous inference (Raphael and Vandin, 2014) is a special case of pathTiMEx, corresponding to the situation when the structure is fixed to a linear path.
However, despite this progress, pathTiMEx still uses a very simplified representation of tumor progression. Future extensions of the model may aim to relax some of its assumptions, such as the hard assignment of genes to pathways, which does not allow for pathway cross talk, or the irreversibility of mutations, which renders our approach not applicable to gene expression data. Moreover, model performance for large datasets with high levels of noise may improve by devising alternative ways of modeling temporal dependencies between waiting times, specifically accounting for false positive and false negative dependencies.
In applications to cancer data, pathTiMEx recapitulates previous knowledge on tumorigenesis, while also offering new insights on the order constraints among pathways in cancer progression. The results of pathTiMEx are highly consistent on the two colorectal cancer datasets analyzed and more consistent with the literature than previous approaches. In glioblastoma, pathTiMEx proposes the existence of a single early causal event consisting of the amplification of CDK4 and the deletion of CDKN2A. These results indicate that pathTiMEx is not only theoretically justified by its treatment of tumorigenesis on the level of pathways as a probabilistic generative process but also successfully applicable in practice.
Footnotes
Acknowledgments
The authors would like to thank Hesam Montazeri for helpful discussions. S.C. was financially supported by the Swiss National Science Foundation (Sinergia project 136247).
Author Disclosure Statement
No competing financial interests exist.
References
1.
AttoliniC.S., ChengY.K., BeroukhimR., et al.2010. A mathematical framework to determine the temporal sequence of somatic genetic events in cancer. Proc. Natl. Acad. Sci. U S A, 107, 17604–17609.
2.
BaburÖ., GönenM., AksoyB.A., et al.2015. Systematic identification of cancer driving signaling pathways based on mutual exclusivity of genomic alterations. Genome Biol. 16, 45.
3.
BeerenwinkelN., ErikssonN., and SturmfelsB.2007. Conjunctive bayesian networks. Bernoulli, 13, 893–909.
4.
BeerenwinkelN., SchwarzR.F., GerstungM., et al.2015. Cancer evolution: Mathematical models and computational inference. Syst. Biol., 64, e1–e25.
5.
BeerenwinkelN., and SullivantS.2009. Markov models for accumulating mutations. Biometrika, 96, 645–661.
6.
BrennanC.W., VerhaakR.G.W., McKennaA., et al.2013. The somatic genomic landscape of glioblastoma. Cell, 155, 462–477.
7.
Cancer Genome Atlas Network, et al.. 2012. Comprehensive molecular portraits of human breast tumours. Nature, 490, 61–70.
8.
Cancer Genome Atlas Research Network. 2011. Integrated genomic analyses of ovarian carcinoma. Nature, 474, 609–615.
9.
CeramiE., GaoJ., DogrusozU., et al.2012. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404.
10.
ChengY.K., BeroukhimR., LevineR.L., et al.2012. A mathematical methodology for determining the temporal order of pathway alterations arising during gliomagenesis. PLoS Comput. Biol. 8, e1002337.
ConstantinescuS., SzczurekE., MohammadiP., et al.2015. TiMEx: A waiting time model for mutually exclusive cancer alterations. Bioinformatics, 32, 968–975.
13.
DempsterA.P., LairdN.M., and RubinD.B.1977. Maximum likelihood from incomplete data via the em algorithm. J. R. Stat. Soc. B, 39, 1–38.
14.
DesperR., JiangF., KallioniemiO.P., et al.1999. Inferring tree models for oncogenesis from comparative genome hybridization data. J. Comput. Biol., 6, 37–51.
15.
Diaz-UriarteR.2015. Identifying restrictions in the order of accumulation of mutations during tumor progression: Effects of passengers, evolutionary models, and sampling. BMC Bioinform. 16, 41.
16.
FarahaniH.S., and LagergrenJ.2013. Learning oncogenetic networks by reducing to mixed integer linear programming. PLoS One, 8, e65773.
17.
FearonE.R.2011. Molecular genetics of colorectal cancer. Ann. Rev. Pathol., 6, 479–507.
18.
FearonE.R., and VogelsteinB.1990. A genetic model for colorectal tumorigenesis. Cell, 61, 759–767.
19.
GerstungM., BaudisM., MochH., et al.2009. Quantifying cancer progression with conjunctive bayesian networks. Bioinformatics, 25, 2809–2815.
20.
GerstungM., ErikssonN., LinJ., et al.2011. The temporal order of genetic and pathway alterations in tumorigenesis. PLoS One, 6, e27136.
21.
HjelmM., HöglundM., and LagergrenJ.2006. New probabilistic network models and algorithms for oncogenesis. J. Comput. Biol., 13, 853–865.
22.
Jerby-ArnonL., PfetzerN., WaldmanY.Y., et al.2014. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell, 158, 1199–1209.
23.
KimY.A., ChoD.Y., DaoP., et al.2015. Memcover: Integrated analysis of mutual exclusivity and functional network reveals dysregulated pathways across multiple cancer types. Bioinformatics, 31, i284–i292.
24.
KirkpatrickS., GelattC.D., VecchiM.P., et al.1983. Optimization by simulated annealing. Science, 220, 671–680.
25.
LawrenceM.S., StojanovP., PolakP., et al.2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature, 499, 214–218.
26.
LeisersonM.D.M., BlokhD., SharanR., et al.2013. Simultaneous identification of multiple driver pathways in cancer. PLoS Comput. Biol. 9, e1003054.
27.
LevandowskyM., and WinterD.1971. Distance between sets. Nature, 234, 34–35.
28.
LoohuisL.O., CaravagnaG., GraudenziA., et al.2014. Inferring tree causal models of cancer progression with probability raising. PLoS One, 9, e108358.
29.
LuoJ., SoliminiN.L., and ElledgeS.J.2009. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell, 136, 823–837.
30.
MadiganD., YorkJ., and AllardD.1995. Bayesian graphical models for discrete data. Int. Stat. Rev., 63, 215–232.
31.
MermelC.H., SchumacherS.E., HillB., et al.2011. Gistic2. 0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41.
32.
RamazzottiD., CaravagnaG., Olde-LoohuisL., et al.2015. Capri: Efficient inference of cancer progression models from cross-sectional data. Bioinformatics. btv296.
33.
RandW.M.1971. Objective criteria for the evaluation of clustering methods. J. Am. Stat. Assoc., 66, 846–850.
34.
RaphaelB.J., and VandinF.2014. Simultaneous inference of cancer pathways and tumor progression from cross-sectional mutation data, 250–264. In Research in Computational Molecular Biology. Springer.
35.
SakoparnigT., and BeerenwinkelN.2012. Efficient sampling for bayesian inference of conjunctive bayesian networks. Bioinformatics, 28, 2318–2324.
36.
ShanmugamC., JhalaN.C., KatkooriV.R., et al.2010. Prognostic value of mucin 4 expression in colorectal adenocarcinomas. Cancer, 116, 3577–3586.
37.
StrattonM.R., CampbellP.J., and FutrealP.A.2009. The cancer genome. Nature, 458, 719–724.
38.
SzczurekE., and BeerenwinkelN.2014. Modeling mutual exclusivity of cancer mutations. PLoS Comput. Biol. 10, e1003503.
39.
TortiD., and TrusolinoL.2011. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol. Med., 3, 623–636.
40.
VandinF., UpfalE., and RaphaelB.J.2012. De novo discovery of mutated driver pathways in cancer. Genome Res. 22, 375–385.
41.
VogelsteinB., PapadopoulosN., VelculescuV.E., et al.2013. Cancer genome landscapes. Science, 339, 1546–1558.
42.
WeinsteinI.B.2002. Addiction to oncogenes—The achilles heal of cancer. Science, 297, 63–64.
43.
WoodL.D., ParsonsD.W., JonesS., et al.2007. The genomic landscapes of human breast and colorectal cancers. Science, 318, 1108–1113.
44.
WuH.T., LeisersonM.D.M., VandinF., et al.2015. Comet: A statistical approach to identify combinations of mutually exclusive alterations in cancer. Cancer Res. 75(15 Suppl.), 1936–1936.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.