
Abstract
Abstract
The successful implementation of the advanced sequencing technology, the next generation sequencing (NGS) motivates scientists from diverse fields of biological research especially from genomics and transcriptomics in generating large genomic data set to make their analysis more robust and come up with strong inference. However, exploiting this huge genomic data set becomes a challenge for the molecular biologists. To corroborate this problem, computational software and hardware are being developed in parallel and become an integral part of life science. While executing the “Genomics project of Indian Drosophila species,” we found strings of Ns in the whole genome sequences generated on Illumina platform. The present article aims at developing a computer algorithm (MATLAB and Python based) for editing raw sequences mainly eliminating bad residues before submitting to the publicly accessible sequence repository. These algorithms will be helpful to life scientists for analyzing large amount of biological data in short span of time.
1. Introduction
W
Data gathering has been fuelled by NGS technology, where huge numbers of primary sequences are generated as dataset in a single experimental run. The high quality datasets are required before proceeding toward the downstream process such as assembly, annotation, single nucleotide polymorphisms identification, and others (Stapley et al., 2010). Therefore, it is necessary to perform preprocessing of these raw sequences generated through NGS technology such as removal of low quality nucleotide sequences, adapters removal, polymerase chain reaction primers and bad residues if any, as they may produce erroneous results. This requires the development of advanced computing softwares and tools for further analysis, and better interpretation of metagenome data (Fernández-Suárez and Birney, 2008; Oliver et al., 2015). Thus, the integration of computational and molecular biologists helps in developing bioinformatics methodologies to deduce meaningful information from these genomic data within short span of time.
Various software tools have been designed for removing ambiguities from large nucleotide sequence datasets such as fasta_clean, Trimmomatic, Adapter Removal, FASTX-Toolkit, SeqTrim, TagCleaner, and so on where they perform trimming and filtering of raw reads (Bolger et al., 2014; Falgueras et al., 2010; Schmieder et al., 2010; Lindgreen, 2012). However, after filtration of all these unwanted sequences, some unknown residues still remain. To clear those away, we have developed an algorithm for editing whole genome sequences (WGS) generated from NGS, which may also be helpful to other researchers dealing with huge genomic data sets.
2. Methods
2.1. Whole genome sequencing
The whole genomes sequences of Indian Drosophila fly were generated using Paired end library on Illumina NextSeq 500 platform using NGS technology (Khanna and Mohanty, 2016). The raw reads were filtered using Trimmomatic v0.30 using optimized parameters. The high quality short reads data obtained after filtering were assembled in form of scaffolds based on de novo approach using CLC Genomics Workbench (version 6.0).
2.2. Algorthims
2.2.1. Sequence editing
Single or multiline gaps in form of strings of Ns of variable length were present either at beginning or end of scaffolds. The scaffolds themselves are of variable length. Manually removing these gaps is a time consuming and error prone task.
The problem of removing these sequences has been formulated as a regular expression based pattern matching problem. Regular expressions are strings of symbols and wildcard characters (*, ?, |, etc.) that define patterns to be searched in an input character sequence (Hopcroft et al., 2006). The regular expressions designed to match for multiline input fasta files divided into scaffolds is as below:
A finite state machine (FSM) has been designed to find these regular expressions in the input file and eventually remove them (Ficara et al., 2008). The FSM can be represented as nondeterministic automata of Figure 1.
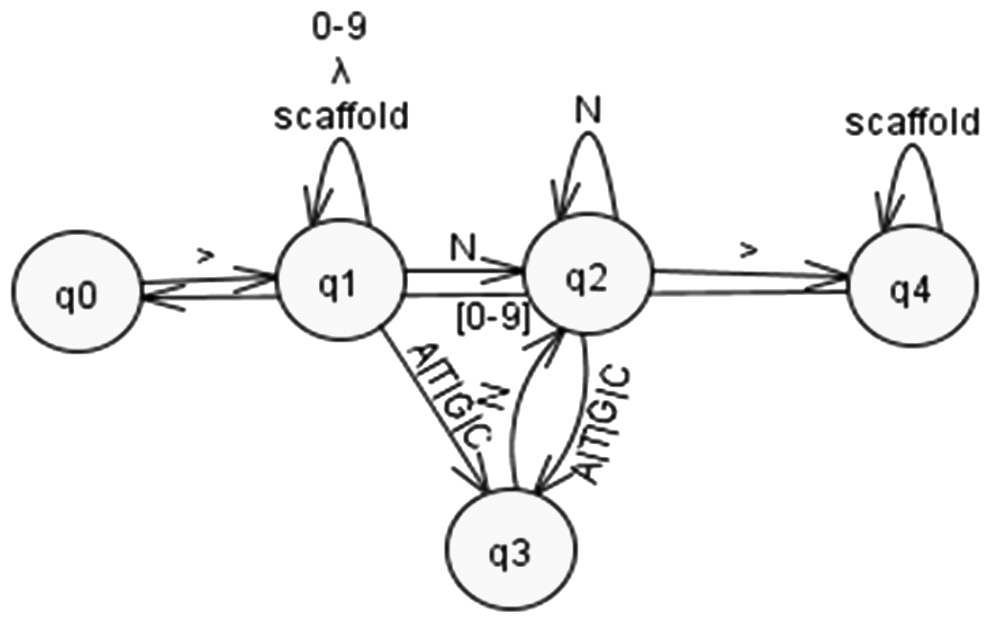
Finite state machine for required pattern recognition.
Sequences recorded from beginning of state q1 to end of q2 are to be deleted for initial gaps removal. Similarly, sequence of q2 recorded just before entering state q4 is to be removed. FSM of Figure 1 has been abstracted into a deterministic program implemented in Python programming language. The step by step details of the method of removal of gaps in each scaffold have been presented as pseudo code in algorithm 1. The scaffold numbers were used as delimiters to mark beginning of a scaffold. The delimiter may vary according to the type of input file.
The algorithm utilizes standard sub sequence finding procedures to find beginning of each scaffold. The trailing genome sequence data were then analyzed for occurrence of prefixes of one or more Ns. These variable length sequences of Ns were then removed from the original fasta file. Identification and removal of gaps at the end of scaffold were computationally more complex than removal at beginning due to repeated back and forth movement in trailing lines of each scaffold. This issue has been addressed in line no 18 of algorithm 1, by initiating traversal of fasta file again, but in reverse order. Algorithm 1 reads and processes each character of the file twice and thus does 2 m operations, where m is the total number of characters in the file. Thus, overall time complexity of search algorithm is still linear, that is, O (|m|). This complexity is comparable to many string matching algorithms proposed for this purpose (Crochemore and Rytter, 2003).
3. Results and Discussion
During Drosophila genome project, we generated WGS of Drosophila species of Indian origin. The high quality reads were assembled into scaffolds following de novo approach using CLC Genomics Workbench. It was observed that some scaffolds contained gaps in forms of strings of Ns of variable length present either at the beginning, middle, or end of the sequence (Figs. 2–4). The gaps present in between the scaffold are of known nucleotide length, which can be substituted later by designing primers and sequencing. Therefore, the single or multiline string of Ns present in the middle of a scaffold need not be removed (highlighted in Figs. 3 and 4). However, the gaps present in the beginning and ends of the scaffold should be eliminated before submitting them to public sequence repository (NCBI). Since the large dataset involved, it was not possible to edit those scaffolds manually. To resolve the problem, the computer algorithm was written in Python and implemented while preprocessing the WGS.

Editing of string of Ns of varying length in the WGS of Drosophila as highlighted.

Editing of string of Ns of varying length in the WGS of Drosophila as highlighted.

Editing of string of Ns of varying length in the WGS of Drosophila as highlighted.
The pseudo code presented in algorithm 1 was implemented in Matlab and Python. The programs were run on two different machines. First was a laptop with core i5 processor and 4 GB RAM. The second machine used was a desktop with core i7 processor and 8 GB RAM. There was a drastic difference in execution time on two machines. With Python-based implementation, it took about one minute to complete the gap removal on laptop, while the same task was done in about one second on desktop. Matlab-based program took about 10 minutes to complete gap removal of one file of size 160 MB. Results of removed gaps in our fasta files are shown in Figures 2–4. The program was also tested for various other genome sequences (Homo sapiens, NCBI accession number: ADDF00000000; Rattus norvegicus, NCBI accession number: AAXM00000000; Mus musculus, NCBI accession number: 000389885.1) retrieved from NCBI database (https://www.ncbi.nlm.nih.gov/). As they are postprocessed sequences, noises (strings of Ns of varying lengths) were generated first and the results of their removal are shown in Figures 5–7. We were able to remove all the required gaps in all the tested files, thus achieving a 100% accuracy rate. Time required for removal using Python on desktop was also found to be of less than 1 second for all files.

Editing of string of Ns in the whole genome sequence of human.
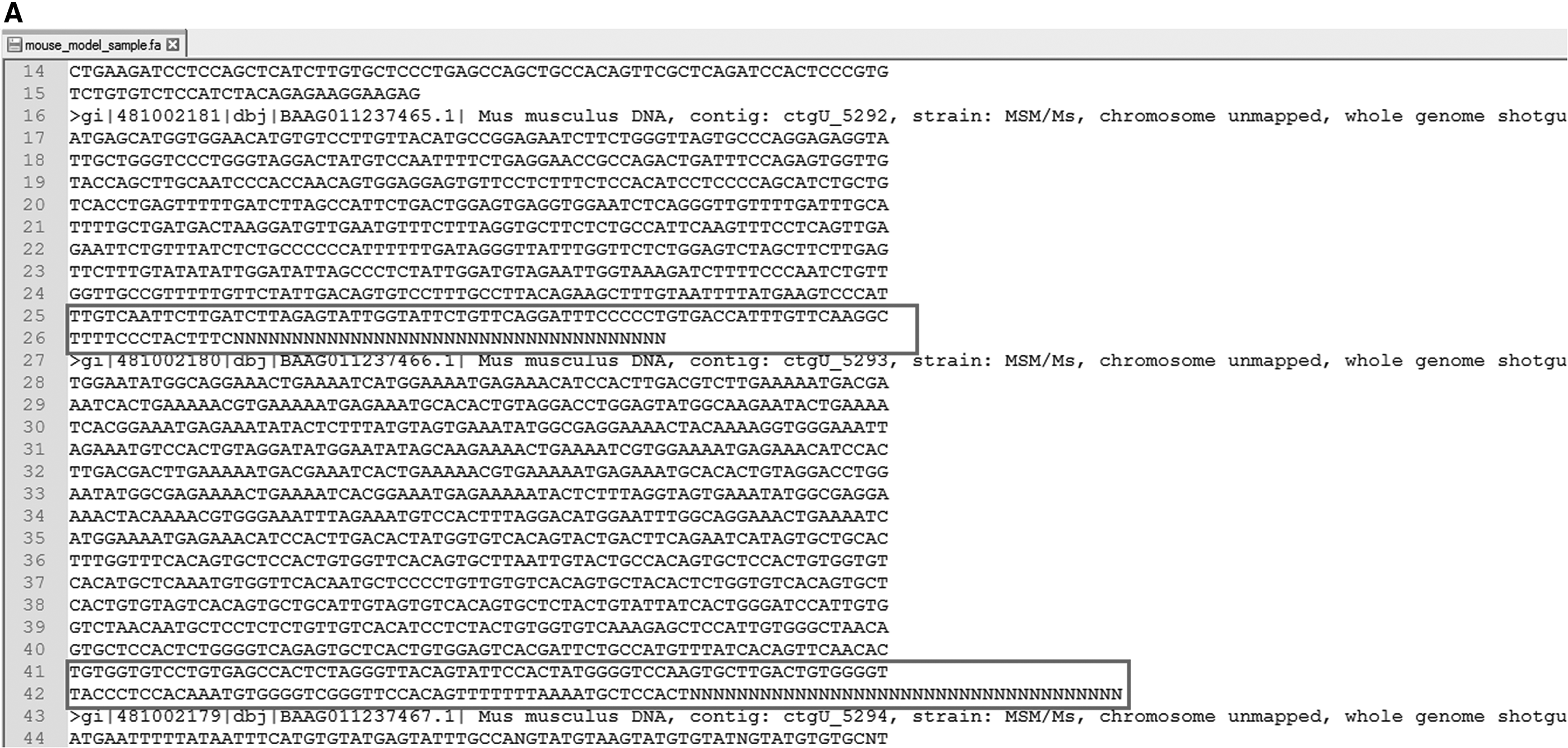
Editing of string of Ns in the whole genome sequence of mouse.
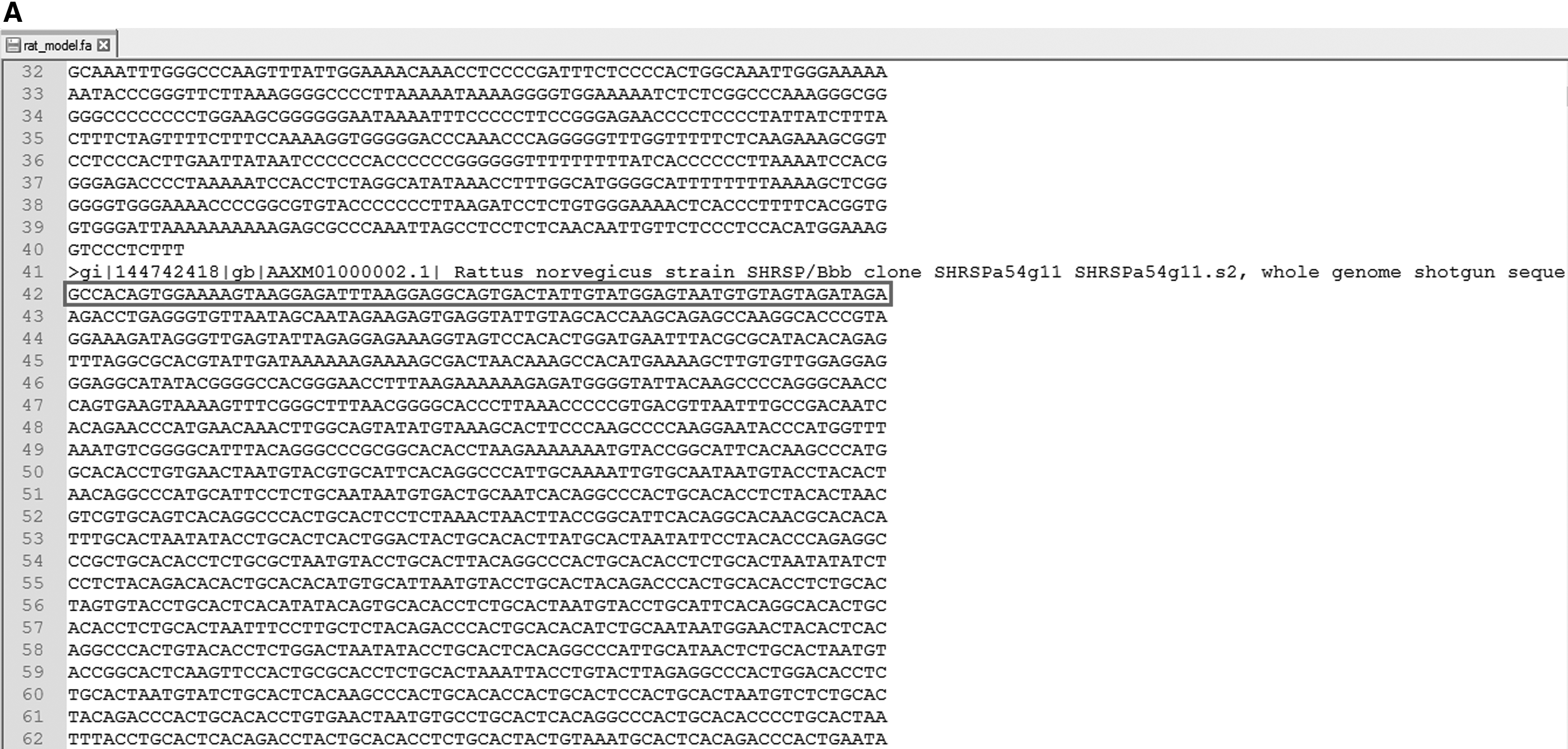
Editing of string of Ns in the whole genome sequence of rat.
There are several other tools that can be used for this purpose. We tried FASTQ/A Clipper tool of FASTX toolkit (http://hannonlab.cshl.edu/fastxtoolkit) (Gordon and Hannon, 2010). It was however, not working in the desired manner and erasing complete lines with valid sequences also. Similar other tools like grep and sed (stream editor) for linux were tried (Crochemore and Rytter, 2003; Abou-Assaleh and Ai, 2004). None of the existing tools were useful in solving the given problem. Hence, this algorithm was developed and it would be useful for researchers working on similar problems.
4. Conclusion
It has become a great challenge in mining large genomic datasets generated through High-Throughput Sequencing technologies and bringing them in an easy-to- understand format, which requires an integration of both genomics and computational biology. Our developed algorithm may be helpful to other researchers while editing WGS.
Footnotes
Acknowledgments
This research was supported by extramural grants from the Department of Science and Technology (DST), India. We would like to acknowledge Jaypee Institute of Information Technology, Noida, India for providing infrastructural support.
Author Disclosure Statement
No competing financial interests exist.