
Abstract
Abstract
The tertiary structure of the proteins determines their functions. Therefore, the predicting of protein's tertiary structure, based on the primary amino acid sequence from long time, is the most important and challenging subject in biochemistry, molecular biology, and biophysics. One of the most popular protein structure prediction methods, called Hydrophobic-Polar (HP) model, is based on the observation that in polar environment hydrophobic amino acids are in the core of the molecule—in contact between them and more polar amino acids are in contact with the polar environment. In this study, we present a new mixed integer programming formulation, exact algorithm, and two heuristic algorithms to solve the protein folding problem stated as a combinatorial optimization problem in a simple cubic lattice. The results from computational runs on a set of benchmarks are favorably compared to known algorithms for solving the 3D lattice HP model as genetic algorithms, ant colony optimization algorithm, and Monte Carlo algorithm.
1. Introduction
P
The determination of the functionality of a protein from its amino acid sequence is one of the fundamental problems in computational biology, molecular biology, biochemistry, and physics. Even experimental determination of these conformations often is difficult and time-consuming. It is common practice for predicting of the tertiary structure on the proteins is to use models that simplify the possible conformations search space. These models reflect the different global characteristics of the protein structure.
The Hydrophobic-Polar (HP) model (Dill and Lau, 1989) describes the protein sequence based on the fact that hydrophobic amino acids must have less contact with water as opposed from the polar amino acids. This leads to the formation of hydrophobic core in the tertiary structure of the proteins.
The optimal conformation of protein folding in the HP model is the one that has maximum number of contacts between hydrophobic amino acids, which gives the lowest energy value (Fig. 1).

Optimal conformation for HP sequence with length 36 amino acids in
It is proved that the protein folding problem in the HP model for 2D and 3D is NP-hard (Berger and Leighton, 1998; Blazewick et al., 2004).
In 2D, the heuristic algorithm described by Traykov et al. (2016) generated folds that are better than the folds obtained by approximate algorithms as Monte Carlo algorithm, Newman's algorithm, Hart–Istrail algorithm, and close to the folds obtained by the Mixed Search algorithm, and Genetic algorithm (Toma and Toma, 1996; Istrail et al., 2000; Chen and Huang, 2005; Istrail and Lam, 2009; Custódio et al., 2004) (Fig. 2).
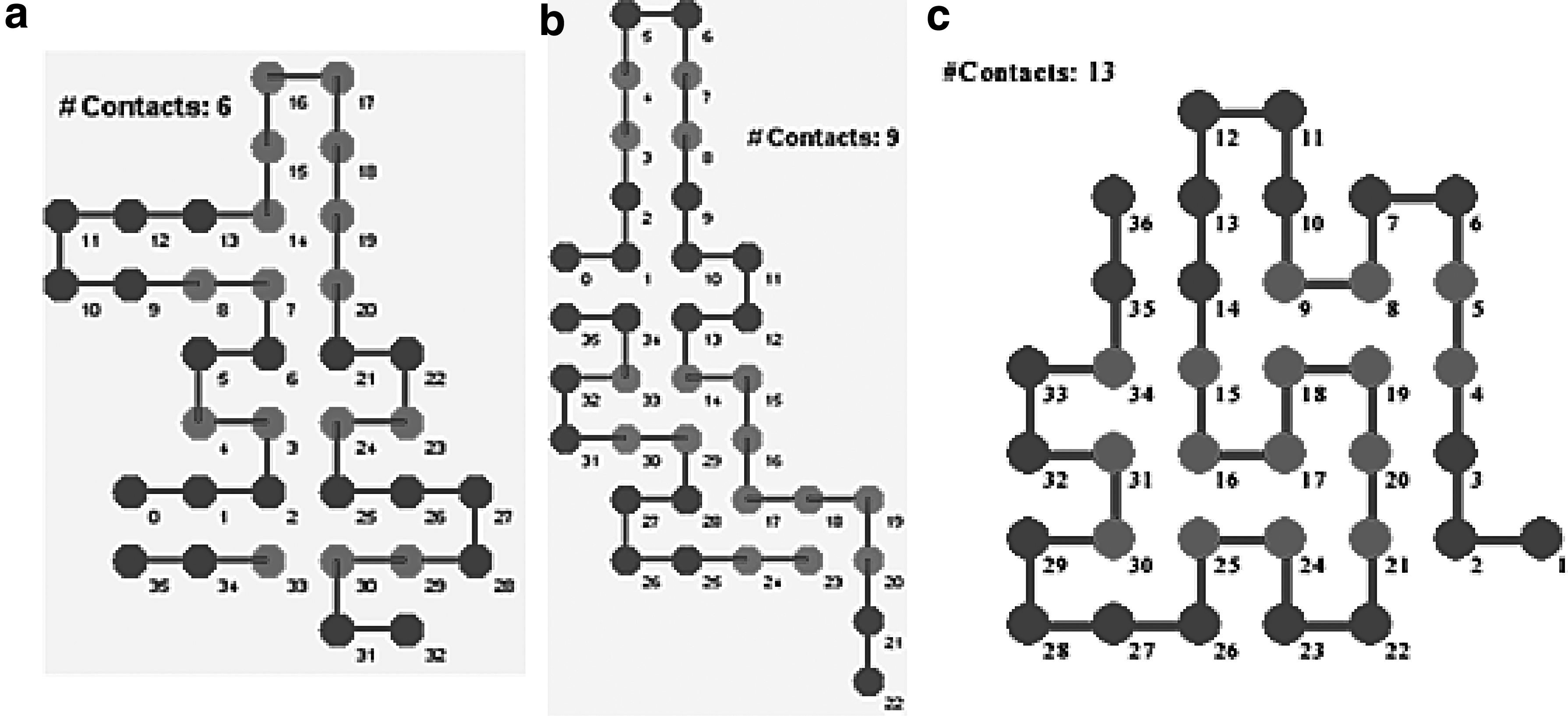
We note that Figure 2a and b are taken from a recommended survey of Istrail and Lam (2009).
The structures above, called self-avoiding paths (walks), are drawn on a square lattice Z2 without overlaps and only hydrophobic interactions are modeled. In the sections below, we (i) formalize the problem, (ii) present it as a problem in so-called alignment graph and give a linear mixed integer programming model, (iii) describe a new heuristics for a cubic lattice case, and (iv) present results from computational runs.
2. Folds in HP Model
The processes, related to the protein folding, are very complex and only a minority of them are explained and understood from the scientists. For this reason, the simplified models, such as Dill's HP model, have become one of the main tools for study of the proteins (Dill and Lau, 1989). The HP model is based on the observation that the hydrophobic interaction between the amino acids is the driving force in the protein folding process. In this model, the 20th amino acids are reduced to two types—H (hydrophobic) and P (hydrophilic). The energy of the conformation is defined as the number of contacts between hydrophobic amino acids (H-H contacts), which are not neighbors in the protein sequence. The optimal conformation is the conformation with lowest energy value, defined as a negative of the number of H-H contacts.
Conformations of the proteins in the HP models are limited to self-avoiding paths in lattice models. The self-avoiding path is a sequence of moves in the lattice, which do not pass through the same position more than once. In a cubic lattice, such a path is simply a sequence of moves from (x, y, z) node to one of the six neighbor nodes. The goal is to find the path that minimizes the energy. Figure 3 shows a schematic representation of a self-avoiding path in a cubic lattice (Zhang and Skolnick, 2004; Yanev et al., 2011).
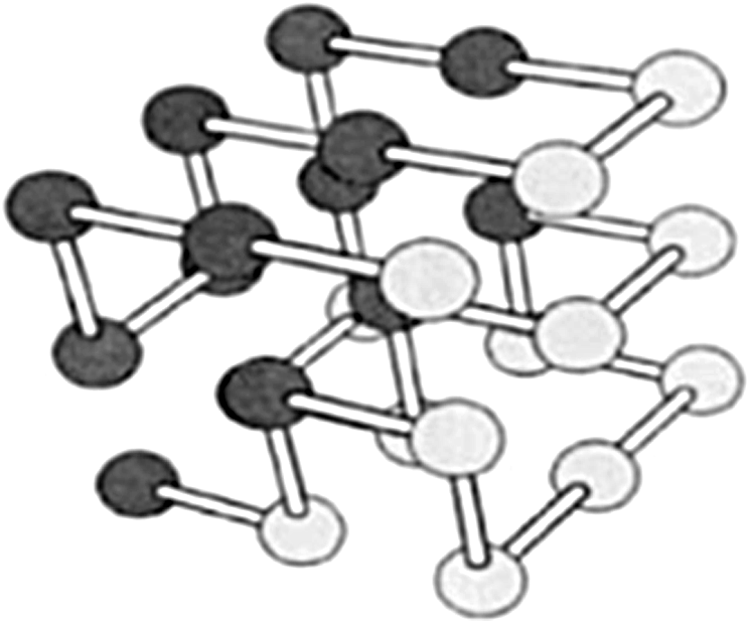
Three-dimensional lattice in the HP model (Thilagavathi and Amudha, 2015).
Briefly, the HP model of protein folding is as follows: given is an amino acid sequence, S = s1, s2, …, sn (sequence of letters over the {H,P} alphabet) and a lattice. The goal is to find conformation of S (to align the letters with a subset of the lattice points) with lowest energy value, that is
Maximize:
The number of H-H contacts
Subject to:
1. Assignment: Each amino acid must occupy one lattice point. 2. Nonoverlapping: No two amino acids may share the same lattice point. 3. Connectivity: Every two amino acids that are consecutive in the protein's sequence must also occupy adjacent lattice points.
For solving the protein folding problem in the HP model, proposed are a number of optimization algorithms, including Evolutionary algorithms (Krasnogor et al., 1998), Monte Carlo algorithms (Liang and Wong, 2001), and Ant-Colony Optimization algorithms (ACOs) (Shmygelska and Hoos, 2005; Thilagavathi and Amudha, 2015; Ramyachitra and Veeralakshmi, 2014).
3. An Integer Programming Formulation
The key ingredients of the problem are as follows: a sub lattice (arbitrary) and a sequence of H, P letters are used below for creating a problem on graphs, which could be solved as an integer programming problem or by means of graph theory only [In the 2D case, the sublattice is a square, and for 3D, a cube with nodes painted in black (set Vb) and white (set Vw) alternately.] Let Gc = (Vc, Ec) be a graph with
Let S be a sequence of n letters on {0,1} alphabet (0-for P, and 1-for H). Let GS = (VS, ES) be a graph associated with S with a node set VS = {1, 2, …, n} and (i, j) ∈ ES if and only if |i − j| ≥ 2 and S[i] = S[j] = 1. Let G = GS ∪ Gc be a complete bipartite graph with node set VS ∪ Vc. The matching (in this case one-to-one mapping of VS to Vc) M = {e1, e2, …, en} with |M| = n is feasible if the covered nodes in Vc define a self-avoiding path. Define function z(ei, ej) = zikjl = 1 if (i, j)∈ES, (k, l)∈Ec, and zikjl = 0 otherwise. Finally, define
Let
Folding problem on graph: For
Converting the HP problem to an optimization problem on graphs allows for building various integer programming models. Most of them involve introducing binary variables, say xik for modeling the feasible matchings from above as 0–1 solutions to simple linear constraints:
Assignment
Nonoverlapping
The objective function could be expressed by linearization of zijkl = xikxjl and/or by partitioning the sum of z in subsums in different ways. We will not present all possible integer programming models, but for the sake of completeness, we add the following constraints to finish modeling the self-avoiding paths:
where n(i) is the set of neighbors of the ith node.
An easily derivable bound to v could be obtained by the following observation, let ODD be the set of odd i, such that S[i] = 1 and EVEN be the set of even i, s.t. S[i] = 1. Since without loss of generality even elements of S are assigned to black nodes and odd elements to the white ones, then zikjl could be equal to 1 only for even–odd couples i, j. Then C2D(S) = 2 × min{|ODD|, |EVEN|} [C3D(S) = 4 × min{|ODD|, |EVEN|} in 3D] is obviously a sharp upper bound to v.
Getting back to the HP folding problem and its conversion to a problem of finding matching that maximizes the number of overlapping edges, one could find a lot of similarity with another problem known as Contact Map Overlap that is (in our case) as follows: for a given two graphs GS, Gc find an embedding (matching) of VS in Vc that maximizes the number of common (overlapped) edges.
A suitable platform for building integer programming models is so called alignment graph G = (Vc ⨂ VS, E) with E = {i, k, j, l}, (i, j)∈Ec, (k, l)∈ES. If we call the arcs in E z arcs then the problem is to find the path in G that activates the maximum number of z arcs. By decomposing the sum (1a), we could obtain different integer programming models like the next one: without loss of generality assume that the set EVEN is of smaller cardinality and they are assigned to Vb. Let yij∈{0,1}, i∈Vb, j∈Vw correspond to the sum of z arcs between rows i and j (Fig. 4). Thus, the problem is to maximize:
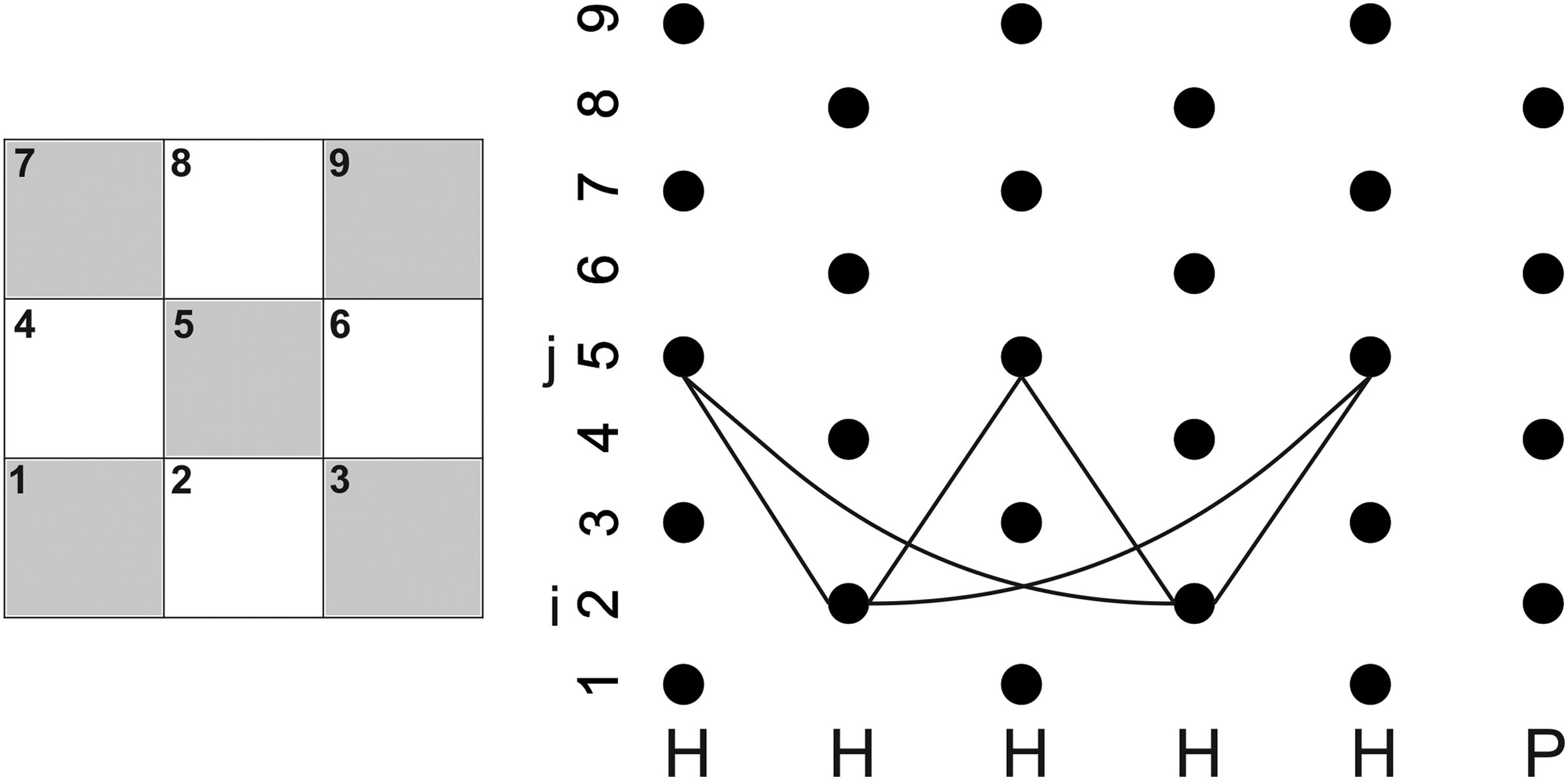
Partial subgraph of alignment-graph G. H = hydrophobic; P = hydrophilic.
subject to additional [to Eqs. (2–4)] constraints allowing yij to be equal to 1:
The partial subgraph of G shown in Figure 4 intends for clarifying the relation of
The model above is a mixed linear integer programming problem with xik binary and using appropriate solvers like CPLEX (www.ibm.com/software/products/en/ibmilogcpleoptistud) or GUROBI (www.gurobi.com/documentation) allows for finding optimal folds for sequences of up to 100 elements on a computer with average capabilities. A challenge for the reader is to prove that the number of binary variables could be reduced from 0.5 to 0.25 nm.
4. Algorithms for Solving the Problem
The main components of the algorithms outlined below are as follows:
• a model builder—creates an lp file for the Model (1–7) • a MIP (mixed integer programming) solver like CPLEX or GUROBI
The common input is a cubic sublattice d1 ≥ x ≥ 0, d2 ≥ y ≥ 0, d3 ≥ z ≥ 0 related to the length n of the input HP sequence S. For the heuristics

The time complexity of these algorithms depends on the segment size, the lattice size, and the computer capabilities. On laptops of average class, both heuristics could find folds of low energy for proteins of any length if the segments length is in [20, 30]. The usage of
5. Computational Experiments
In this chapter, we compare our algorithms with Genetic algorithm, ACO, and Evolutionary algorithm with Backtracking in 3D lattice. For computational experiments, we use eight HP sequences that are known in the literature benchmarks for 3D lattice in the HP model (Jiang et al., 2003; Garza-Fabre et al., 2003) and one additional HP sequence with length 102 amino acids, which is the benchmark sequence that we propose (Table 1).
The symbols H i , P i and (…) i in Table 1 show i repeats of character or sequence.
In Table 2, we compare the obtained results by our algorithms (the columns
ACO, Ant-Colony Optimization algorithm; BKS, best known solution; EA, Exact algorithm; GA, Genetic algorithm; HE-1, Heuristic algorithm-1; HE-2, Heuristic algorithm-2.
The protein sequence for which we improve the BKS.
The column
For sequences with length 48, 50, and 60 amino acids (Fig. 7), the algorithm

Solution for protein sequence with length 102 amino acids.
Figures 6 and 7 show the obtained conformations for protein sequences with length 102 and 60 amino acids.

Protein folds with length 102 amino acids (53 contacts). Light = hydrophobic; dark = hydrophilic.
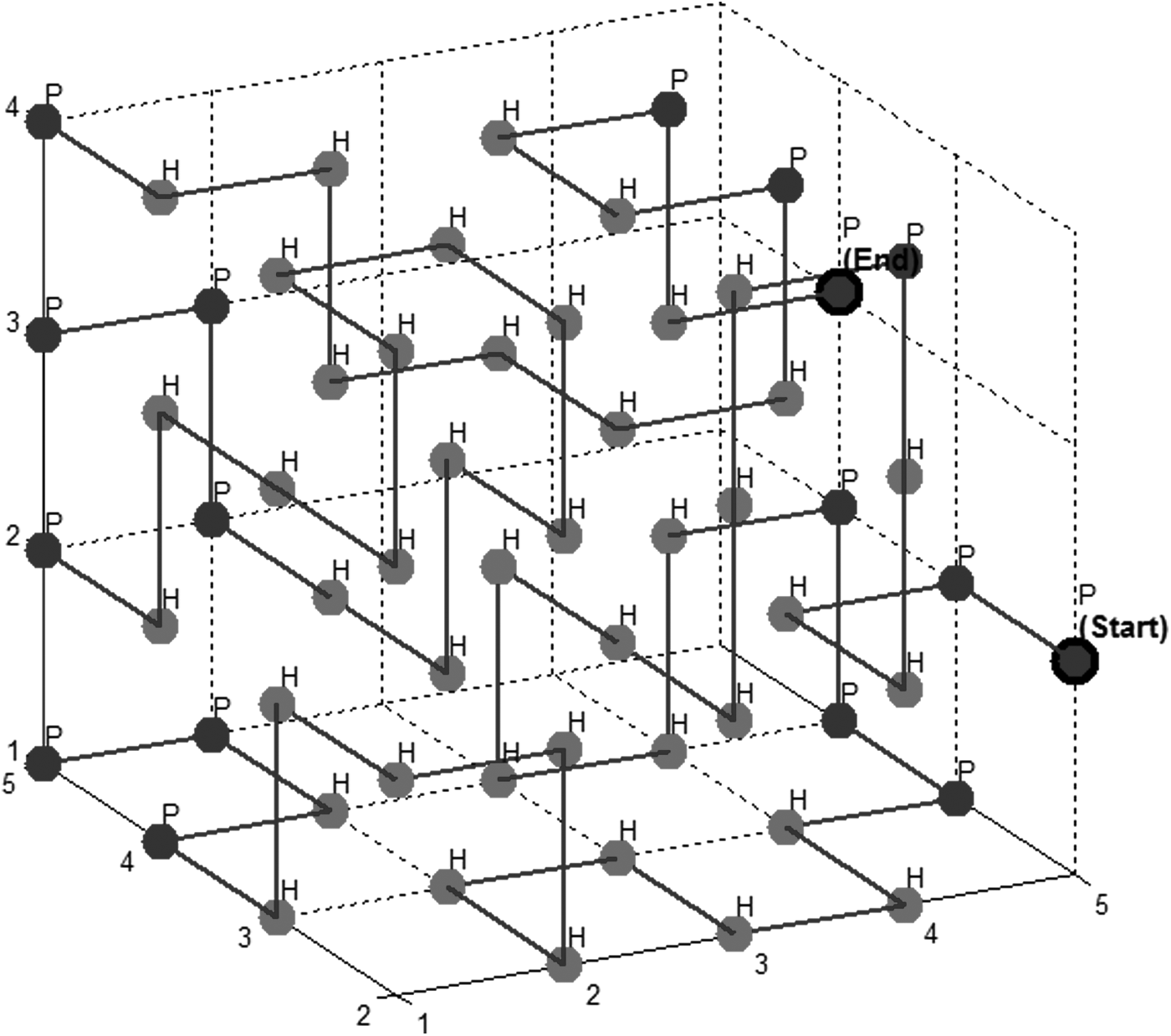
Protein folds with length 60 amino acids (55 contacts). Light = hydrophobic; dark = hydrophilic.
The machine that we use for realization of the computational experiments is a laptop with Intel(R) Core(TM) i7-3632QM (2.20 GHz, 6 MB cache) processor and 8 GB RAM.
6. Conclusion
A novel mixed integer programming model for the HP folding problem is proposed, which is successfully used in an exact algorithm for the case of cubic lattices and also as a component for efficient heuristics. Without any further improvements, the exact algorithm is capable for finding (by use of solvers like CPLEX; www.ibm.com/software/products/en/ibmilogcpleoptistud) the optimal number of contacts for sequences of lengths up to 100 HP characters. This limit is overcome by the heuristics, which (at least over the available benchmarks) create near optimal folds.
Footnotes
Acknowledgments
This work is partially supported by the project of the Bulgarian National Science Fund, entitled: “Bioinformatics research: protein folding, docking and prediction of biological activity,” code NSF I02/16, 12.12.14.
Author Disclosure Statement
No competing financial interests exist.