
Abstract
Abstract
1. Introduction
T
Many of the works in it were conducted by experimentally analyzing abnormal phenotypes from silencing a particular gene. Hamahashi et al. (2007) presented a system that automatically measures the cell division pattern of Caenorhabditis elegans. They also statistically analyzed two inferences on the spatial arrangement of cells and the span between the end of the four-cell stage and the beginning of the eight-cell stage of C. elegans for both par-1 mutation embryos and wild-type (WT) embryos. However, the former way only provides faint information for identifying concrete gene functions, and the latter method is not only arduous but also restrained. The first limitation of it is that we usually need a large number of RNAi and WT embryos to reach a statistical result. The second limitation lies in the efficiency. To verify whether a certain gene has a presupposed function or not, the common method is selecting features according to the function and statistically testing the WT and RNAi embryos, which is problem oriented and only applicable for a predetermined problem related to a particular gene function. Therefore, its efficiency is limited in large-scale gene analysis.
In this article, we propose a WT-based data-driven method to study the concrete function of each gene associated with a specific category of abnormal phenotypes. It firstly determines the recognized features based on a certain phenotype and all the involved genes. Then, after merging these features using principal component analysis (PCA), statistical models are built based on a large number of WT embryos. Finally, different RNAi embryos corresponding to all the involved genes are screened with the built statistical models. The advantages of this approach are as follows: first, it can be applied to genes with a small number of RNAi embryos; second, a number of genes can be analyzed using the same models; and third, it is not necessary to presuppose the particular function of a gene, instead its function is confirmed by the statistical testing results of the built models. To evaluate the effectiveness of the framework, we choose the phenotype, P1/AB asynchrony of divisions, as an example.
The nematode C. elegans is one of few animals in which essential embryonic genes have been identified through genomewide RNAi screening (Fraser et al., 2000). It also has a fixed cell lineage and a precise cell fate map (Sulston et al., 1983), which makes it a perfect animal to functional gene analysis.
Cell division of C. elegans is asymmetric, generating the two daughter cells with different sizes, which plays an important role in the generation of cellular diversity during development (Horvitz and Herskowitz, 1992; Guo and Kemphues, 1995). The asymmetry of division is crucial not only to C. elegans but also to Drosophila melanogaster and mouse (Betschinger and Knoblich, 2004; Gönczy, 2008).
Usually, for the first two blastomeres of a WT embryo, the anterior blastomere (AB) is arguably larger than the posterior blastomere (P1) (Arata et al., 2015). Kemphues et al. (1988) verified that silencing par-1, par-2, and par-3 might disturb the normally asymmetric cleavage and generate variant daughter cells. More relevant analysis can also be seen in Hara et al. (2013) and Ladouceur et al. (2015). In Phenobank (Sönnichsen et al., 2005), currently known genes with the similar function that may influence the first round of cell division (from P0 to P1 and AB) are compiled in the category of “P1/AB Asynchrony of Divisions.” Although these genes have been found and listed in Phenobank, their, respectively, specific functions on the first round of cellular division remain unclear. The asynchrony of division may express in multiple facets, such as the difference of lifetime, size, growth ratio, or geometric shape. Understanding the specific function of these genes is valuable to finally comprehend the whole gene expression regulatory network. Because the proposed method requires a large number of WT embryos to carry out the analysis, we use Worm Developmental Dynamics Database (WDDD) as the database. WDDD includes the records of 136 RNAi and 50 WT embryos developing from 1 to 16 cells (Kyoda et al., 2013), which makes it a desired platform (see Section 3.1. for details) for our study.
2. Methods
2.1. Data
Using four-dimensional (4D) differential interference contrast (DIC) microscopy, WDDD has 50 sets of quantitative data from WT embryos. After silencing embryonic genes on chromosome III individually, 72 genes with 136 sets of quantitative data from RNAi experiments were also built (Kyoda et al., 2013; http://so.qbic.riken.jp/wddd). For each set, there are 180 time points (40 seconds/time point), and a three-dimensional (3D) image is stored at each time point. In each 3D image, the z-stack includes 66 focal planes (0.5 μm/plane) and there are 600 × 600 pixels (0.105 μm/pixel) in each focal plane. Besides 4D DIC images, WDDD also provides the dynamic coordinates of outlines of each nuclear region during the first three rounds of division and their corresponding blastomere names. In the 72 genes, 6 of them are categorized into “P1/AB Asynchrony of Divisions” in the Phenobank. They are dcn-1, ket-754, mcm-5, par-2, par-3, and rnr-1.
After checking the number of time points of P1 and AB for the 136 embryos, we discard deficient embryos for two reasons as follows: (1) the P1 or AB dies (without daughters) and (2) the number of time points of P1 (or AB) is less than 3 (therefore, the computed features may be unreliable.) Among the discarded embryos, T23G5.1_061102_01 belongs to rnr-1. Thus, we use the remains of five genes. Its corresponding RNAi embryos are shown in Table 1.
2.2. The Algorithm
According to the introduction, we can build the
The algorithm of gene function analysis

2.3. Features
2.3.1. Choosing features
A nucleus of P1 is arguably smaller and more slender than AB. Therefore, we use four well-known features: volume, surface, diameter, and compactness to measure. The first three characteristics are related to size, whereas the last one reflects the level of slenderness. Although there are many shape characteristics being proposed (Loncaric, 1998; Zhang and Lu, 2004), the four characteristics used in this study are both easy to compute and well recognized. For each embryo, there are many time points (usually 15–40) and corresponding nuclei at the stage of P1 (or AB). For the purpose of condensing data, we use mean value and standard deviation (std) in the period of P1 (or AB) to reflect the morphological characteristics.
The definition of diameter in 3D space used in this article is a natural generalization of the definition in two-dimensional (2D) space. We call the distance between two parallel tangent planes of an object as diameter. It is also known as Feret diameters (Glasbey and Horgan, 1995). Along one direction in 3D space we can calculate one diameter (Fig. 1). Therefore, if we choose 1000 directions in 3D space, 1000 diameters will be worked out. For a nucleus at certain time point, we can compute only one value for volume or surface, whereas for diameter, the number of computed values is 1000 if we use 1000 directions. Based on these values, we compute the mean value to reflect their size and the ratio of the minimal value to the maximal value (min/max) to reflect their compactness. After that, we can calculate the mean value and std value for an embryo on the whole time points within the period of P1 (or AB). In summary, we use 10 features (Table 2) to analyze.
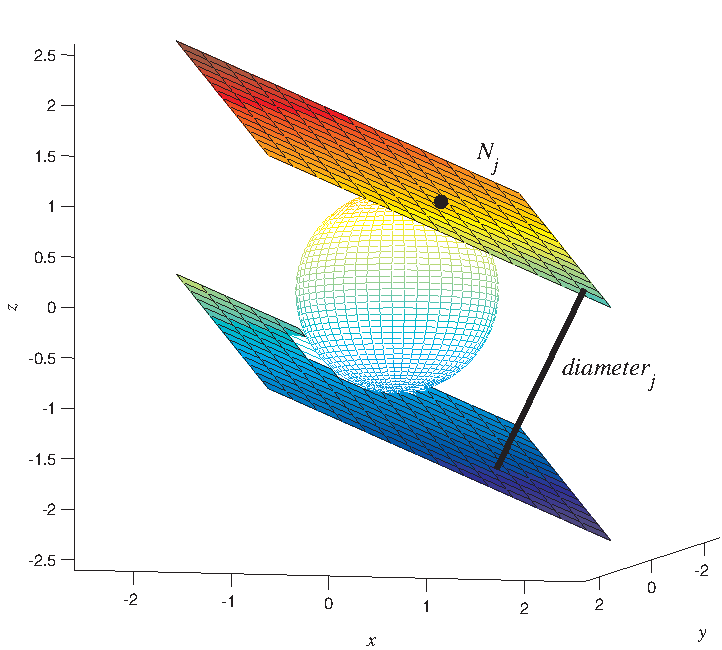
Feret diameter. x, y, and z are the three orthogonal axes in euclidean space.
2.3.2. The computation of the features
WDDD provides the coordinates of outlines for each nuclear region. To simplify the computation, we use the number of involved voxels to represent volume and surface area. Rosenfeld pointed out that using this method to compute some classic shape characteristics such as perimeter may cause inconsistent results and give theoretical analysis (Rosenfeld, 1974). For the data in WDDD, however, because the images are built in the same way under the same condition, if we process the data of different embryos in the same way, the influence of discretization is small and will not bring meaningful errors.
2.3.2.1. Volume
For the nucleus at t (t = 1,
2.3.2.2. Surface area
For the nucleus at t (t = 1,
2.3.2.3. Diameter
For the nucleus at t (t = 1,
According to the meaning of diameter in this article, we use Golden Section Spiral (GSS) method (Treeby and Cox, 2010) to obtain evenly scattered Nd points
between Nd and the central angle θspace of two adjacent points Qd, Qd+1 (Fig. 3). The computation burden and the computation accuracy are a trade-off. In this study, we set Nd = 1000, which makes the θspace less than 10°.
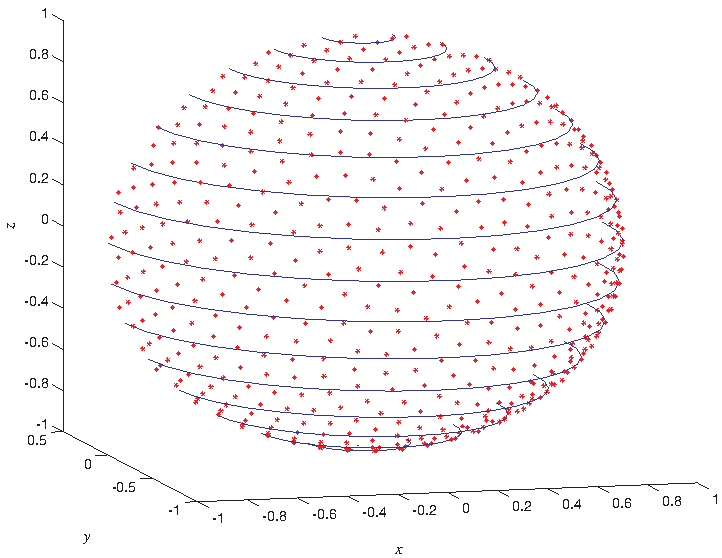
The generated points in a half sphere by GSS; x, y, and z are three orthogonal axes in euclidean space. GSS, golden section spiral.
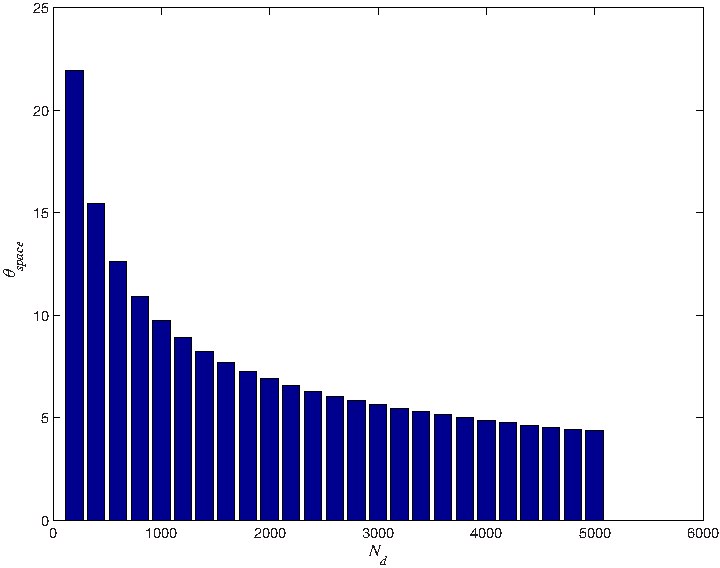
The relationship between Nd and the angle interval. x-axis represents Nd, and y-axis is the angle interval.
We use two steps to calculate the features related to diameter. (1) The computation of diameter. For the nucleus at t (t = 1,…,T) time point of an AB, using:
(2) Compute the features related to diameter. The equations are:
Then, we can compute
2.3.2.4. Compactness
The computation of compactness (Attneave and Arnoult, 1956) is based on Young et al. (1974). For the nucleus at t (t = 1,…,T) time point of an AB, based on the computed VtAB and StAB, we can derive:
The two features
2.3.3. The final built features
2.3.3.1. Normalizing magnitude
After computing the features related to volume, surface, diameter, and compactness, we adjust their magnitudes to keep them in line with the length. After standardizing, the final used features are (the features of AB are similar):
2.3.3.2. Building features
The standardized data have removed the influence of magnitude. However, there are two values (belongs to P1 and AB, respectively) corresponding to the identical feature of the same embryo, which is hard to handle when testing or evaluating an embryo. Therefore, suppose the 50 values of FiP1 (i = 1,…,NF, NF = 10) are
2.4. Building models
To compile similar geometric characteristics from these features, we use PCA for feature selection and feature fusion. To identify the degree of abnormality for each RNAi embryo, the first four principal components of WT embryos are used to build normal models. The reason of choosing four components in our case is based on the ratio of cumulative sum of eigenvalues; in this study we use 80% (Fig. 4). Experimental results show that each of the first four components has a specific physical meaning, which will be discussed in Section 4.
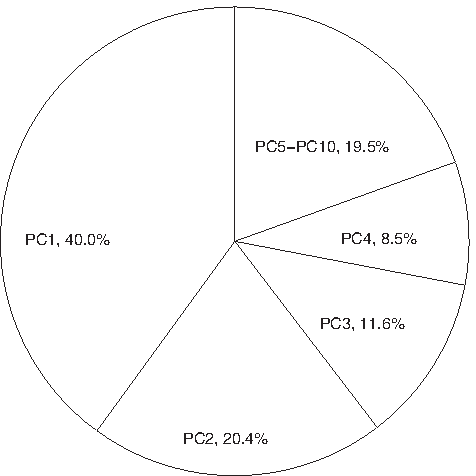
The eigenvalues of PCA. PCi (i = 1,…,10) is the ith eigenvalue. PCA, principal component analysis.
For each of the four eigenvectors em (d1 ≥ d2 ≥ d3 ≥ d4 ≥ dk, k = 5,…, NF, m = 1, 2, 3, 4), we can project
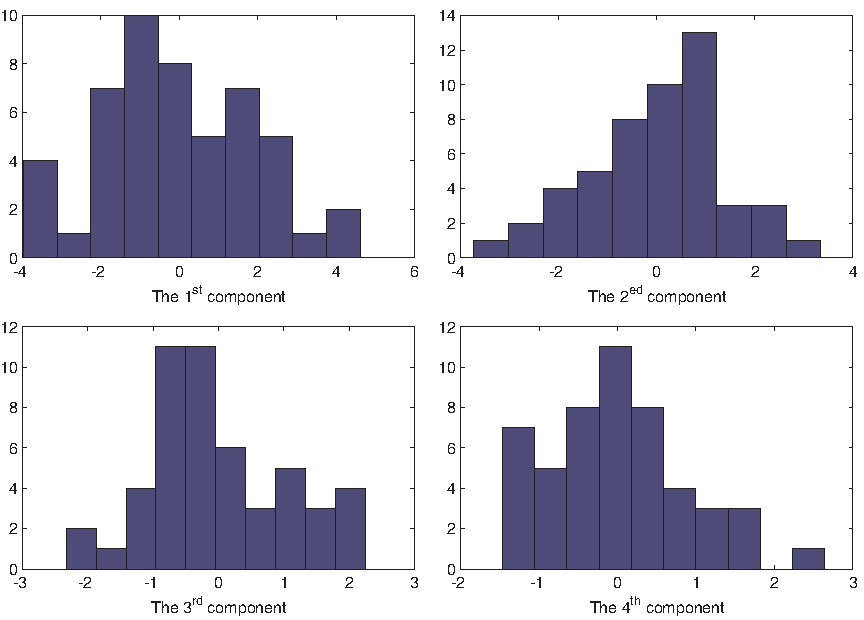
The histogram of projected NWT WT embryos on different components. WT, wild type.
2.5. Abnormal RNAi detection
The identification of abnormal RNAi embryos is based on the normal models built from WT embryos. First, the value of features Fi,j (i = 1,…,NF, j = 1,…, NRNAi, NF = 10, NRNAi = 136) for RNAi embryos is calculated based on the computed μmWT, σmWT, and em from WT embryos with the same way that WT embryos were treated. Second, we use Z-test according to N(μmWT, σmWT) and set p value as 0.05 and 0.1 to test abnormal embryos.
3. Results
The computed eigenvalues and eigenvectors in PCA are listed in Figure 4 and Table 3, respectively. According to the PCA result, the first, second, third, and fourth component, respectively, capture 40.0%, 20.4%, 11.6%, and 8.5% of the variance across all features and totally cover more than 80% of the variance. Choosing the three largest weights for the first four eigenvectors and modifying by the limitation that only one eigenvector could be assigned for a feature, the result shows that eigV1 (the eigenvector corresponding to the first eigenvalue, similarly hereinafter) mainly represents the first, third, and fifth features, which are volume mean, surface mean, diameter mean; eigV2 mainly represents the second, fourth, and sixth features, which are volume std, surface std, and diameter mean std; eigV3 mainly represents the seventh, ninth, and tenth features, which are diameter min/max mean, compactness mean, and compactness std; and eigV4 mainly represents the eighth feature, which is diameter min/max std. According to the physical meaning for each feature, it seems that the first component has strong correlation to the difference between P1 and AB on the scale or size of a nucleus. In contrast, the second component is mainly related to the variation of scale or size of a nucleus along different time points. It may reflect the difference of development between P1 and AB. As for the third component, it roughly reflects the geometric shape of a nucleus. Finally, the fourth component, it seems to associate with the variation of shape of a nucleus along different time points.
eigV1–eigV4 represent the first four eigenvectors, and F1–F10 represent the NF features.
After building four normal models by each of the first four components, we can use the four built models to detect RNAi embryos. For these embryos, the detection results are summarized in Table 4 and Figure 6. In Figure 6, we show the result of identification based on each of the component. To make the figures more understandable, when showing the detection result of the ith (i = 1, 2, 3, 4) component, we plot a 2D figure with ith component as x-axis and the probabilities of the value for the corresponding model as y-axis. From the result, we can draw the following conclusions:
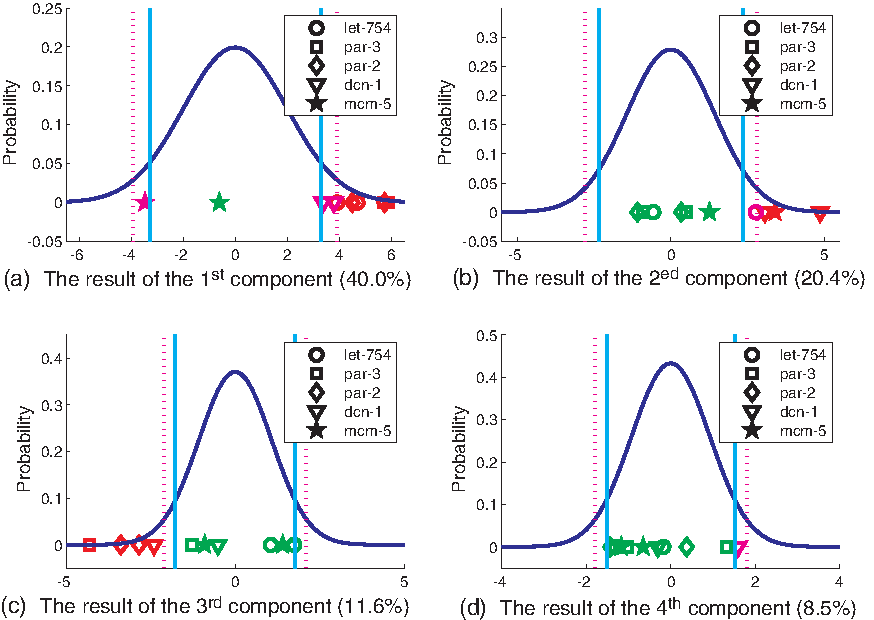
Projected 10 RNAi embryos of the five known genes (Table 1) on the ith components (i = 1, 2, 3, 4). x-Axis is the projected value and y-axis is the probability of the projected value based on the corresponding built normal distribution. The cyan solid lines and the pink dotted lines are the decision borders when p value is 0.10 and 0.05, respectively. Red objects are the projected values of embryos that are identified as abnormal cases when p value is 0.05. Pink objects are the projected values of embryos that are identified as normal cases when p value is 0.05, whereas abnormal cases when p value is 0.10. Green objects are the projected values of embryos that are identified as normal cases when p value is 0.10.
CDF1–CDF4 represent the cumulative distribution function of testing for an embryo when m is 1, 2, 3, and 4. Bold values correspond to abnormal embryos, which are detected when p value is 0.05, and values with an italic font correspond to abnormal embryos when p value is 0.1.
First, let-754, par-2, and par-3 play important roles in the asymmetric division on scale, because silencing one of them tends to change the difference of P1 and AB on scale. The knockdown of dcn-1 by RNAi arguably changes the division similar to the former three genes, whereas the knockdown of mcm-5 arguably changes the division in an opposite way.
Second, the genes, dcn-1 and mcm-5, are crucial in the asymmetric division on the change of scale (we guess it is related to the growth ratio), because silencing one of them tends to change the difference of P1 and AB on the variation of scale or size of a nucleus along different time points. The gene, let-754, arguably has the same function, whereas par-2 and par-3 have negligible influence on the variation of scale.
Third, dcn-1, par-2, and par-3 serve important functions in the asymmetric division on geometric shape, because silencing one of them tends to change the difference of P1 and AB on geometric shape.
4. Conclusions
We propose a framework to identify gene functions in this article. To validate the framework, we apply it to identify the concrete functions of five known genes appearing both in Phenobank and in WDDD, which are critical in the asymmetric division of C. elegans on the first round. Experimental results show the effectiveness of this framework in identifying specific gene functions for certain category of abnormal phenotypes. Because the scheme introduced in this study can be applied to other genes making only few modifications, it may have extensive applications. One requirement of the method is that it needs a large number of WT embryos to build reliable models.
Footnotes
Acknowledgments
This work was supported, in part, by the National Bioscience Database Center (NBDC) of the Japan Science and Technology Agency (JST), the Strategic Programs for R&D (President's Discretionary Fund) of RIKEN, and the Grant-in-aid for Scientific Research from the Japanese Ministry for Education, Science, Culture, and Sports (MEXT) under the Grant No. 16H01436.
Author Disclosure Statement
No competing financial interests exist.