
Abstract
Abstract
1. Introduction
M
Although dynamic programming like Needleman–Wunsch algorithm (Needleman and Wunsch, 1970) can be generalized in theory to produce alignments for any number of sequences, unfortunately, this leads to an explosive increase in computer time and memory requirements as the number of sequences increases (Taylor, 1990). MSA remains under continuous development, and is regarded as one of the most challenging problems in the field of bioinformatics and computational biology (Chatzou et al., 2016). Furthermore, the computation of an accurate MSA has long been known to be an NP (nondeterministic polynomial time)-complete problem (Wang and Jiang, 1994), a situation that explains why >100 alternative methods have been developed these last three decades, and many mainstream methods use heuristic and combinatorial optimization algorithms for obtaining approximation of optimal solution, imposed by the NP-complete nature of the problem (Chatzou et al., 2016).
We develop a new method that performs pairwise alignment in O(n) time between two highly similar sequences based on a suffix tree structure, which is well known for its string processing power. Here, we take two sequences that have at least 56.8% nucleotides in common as highly similar sequences. The number of nucleotides (56.8%) is calculated in the experiment sector. We also design a novel MSA method called MASC (multiple sequence alignment based on a suffix tree and center-star strategy), which has extremely high performance and accuracy with our new pairwise-alignment algorithm. Furthermore, we implement MASC on the Spark-distributed parallel framework for use case for massive scale sequence data and accelerate the method by using multiple computing node parallel programming.
2. Related Work
MSA one of the most widely used modeling methods in biology is indeed an important modeling tool whose development has required addressing a very complex combination of computational and biological problems (Chatzou et al., 2016).
The most commonly used heuristic methods involve progressive-alignment strategy (Hogeweg and Hesper, 1984), iterative-alignment strategy (Wallace et al., 2005), or center-star strategy (Zou et al., 2009). ClustalW (Thompson et al., 1994) is the most widely used implementation of the progressive-alignment strategy. MAFFT (Lounkine et al., 2012) is quite fast, using a fast Fourier transform algorithm along with a progressive-alignment strategy.
Progressive alignment generates a quasi-phylogenetic guide tree among the sequences and gradually builds up the alignment in a pairwise manner, following the order provided by the tree. Although successful for a wide variety of cases, the main caveat of the progressive-alignment approach is the existence of local minima, for example, the early computation of the first pairwise alignment may prevent the computation of a globally optimal MSA. Errors made in initial alignments cannot be rectified later as the remainder of the sequences are added (Notredame et al., 2000). Apart from this, progressive alignment is time consuming when dealing with large-scale data sets, due to its nonlinear time complexity (Zou et al., 2012).
The iterative strategy (Wallace et al., 2005) is an interesting alternative method, which is based on tree-based progressive strategy and involves re-estimating trees and alignments until both converge. Iterative strategies do not provide any guarantee of an optimal solution, but are reasonably robust, and are much less sensitive to the number of sequences than their deterministic counterparts (Gotoh, 1996). PRRN (Gotoh, 1996) and MUSCLE (Edgar, 2004) employ an iterative-alignment strategy.
The center-star-alignment strategy, which is utilized in our project, is a fast method for solving MSA, and suitable for highly similar sequences. Assume that it takes O(t) to run a pairwise alignment between two sequences. The complexity of center-star strategy is O(
The main approach underlying the center-star method is to transform MSA into pairwise alignments based on a “center sequence.” Center sequence is selected using a similarity matrix, a symmetric matrix that stores the similarity scores of each of the two sequences. The similarity scores are calculated by pairwise alignments, which indicate the similarity of two sequences. The construction of this matrix takes O(
Regardless of which heuristic method is used, the main simplification idea common to all is to reduce the MSA to a series of pairwise sequence alignments. Consequently, pairwise alignment is a dominant component of all MSA techniques. Traditional dynamic programming algorithms, such as Smith–Waterman (Smith and Waterman, 1981) and Needleman–Wunsch (Needleman and Wunsch, 1970), require O(n2) temporal and spatial complexity to perform pairwise alignment, where n is the maximum length of two sequences. Other faster algorithms have been developed, such as MAFFT (Lounkine et al., 2012), which is O(nlogn). These algorithms work extremely well on conventional tasks with multiple single protein sequences, cDNA sequences, or relatively short genomic DNA sequences containing a single gene and simple intron interruptions. Furthermore, when performing the pairwise alignment between two very long sequences, such as mtDNA (Iborra et al., 2004), or whole genomes, many algorithms either run out of memory or take too long to complete (Delcher et al., 1999). To the best of our knowledge, there is no efficient and effective pairwise-alignment method that requires O(n) time complexity.
3. Algorithms
We contribute three novel points for MASC. First, suffix tree (Ukkonen, 1995) is employed for pairwise alignment, which can reduce the time complexity to O(n) for similar DNA sequences. Here n is the sequence length. Second, we improve the center-star MSA strategy by selecting randomly center-star sequence (Zou et al., 2012). Experiments in Section 4.2 prove that center-star sequence selection would not influence the performance for similar DNA sequences. Finally, MASC is implemented on the Spark-distributed parallel framework pursuing further acceleration by parallel computing (Zaharia et al., 2010).
3.1. Suffix tree pairwise alignment
The basis of our method is a data structure known as a suffix tree, which is a compressed tree (Aho and Corasick, 1975) containing all the suffixes of a given text as keys, and positions in the text as values. Suffix trees allow particularly fast implementations of many important string operations (Baeza-Yates and Gonnet, 1996). The suffix tree for the string S of length n is defined as follows:
The tree has exactly n + 1 leaves numbered from 0 to n. Except for the root, every internal node has at least two children. Each edge is labeled with a nonempty substring of S. No two edges starting out of a node can have string labels beginning with the same character. The string obtained by concatenating all the string labels found on the path from the root to leaf i spells out a suffix, a simple substring that begins at any position in S and extends to the end of S.
In Figure 2, a complete suffix tree of string “agctggcc$” is shown.
The efficient algorithms given by Ukkonen can construct a suffix tree in linear time with O(n) computational space from a string, where n is the length of the string (Ukkonen, 1995). Ukkonen's algorithm also reduces suffix tree construction to O(n) time, for constant-size alphabets, and O(nlogn) in general. Within a bioinformatics context, the tree alphabets consist of {A, G, C, T} or {A, G, C, U} for DNA or RNA, respectively, or the 20 amino acid symbols for proteins. Therefore, the time cost is linear for all molecular biological sequences (Barsky et al., 2008).
The inputs to the pairwise alignment are two sequences. We call them sequence S1 and sequence S2 for convenience, it is assumed that the sequences to be compared are highly similar. Therefore, there are many common substrings in both S1 and S2. The alignment process consists of four steps, which are shown in Figure 1.
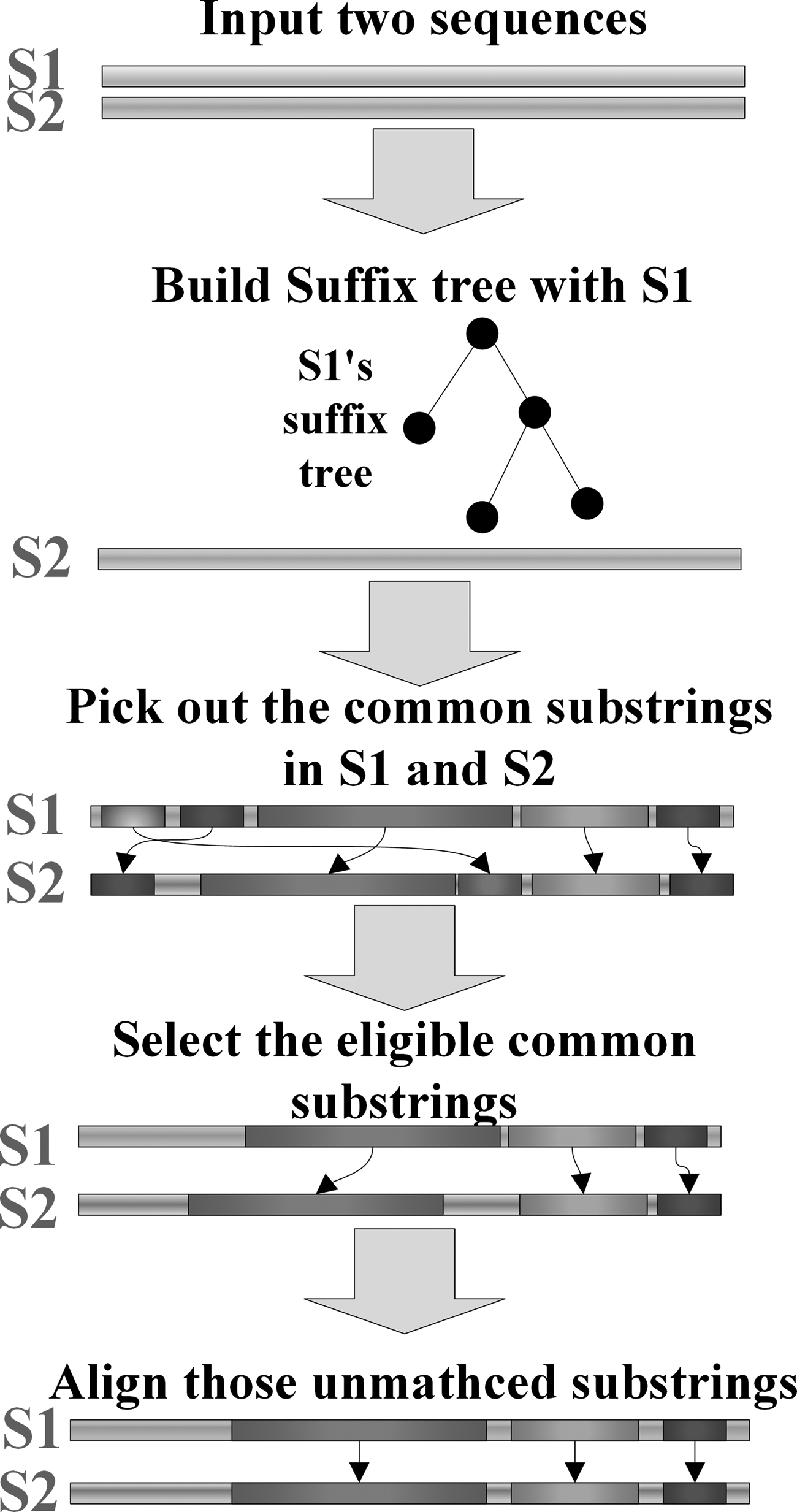
Pairwise-alignment process using a suffix tree.
1. The first step of our method is to build a suffix tree from one sequence. Here S1 is assumed as the chosen one, and the tree's name is tree-S1. The tree construction consumes O(n) time with Ukkonen's algorithm, n is the length of S1.
2. Then we use a process to pick out common strings. The pseudocode of the process is shown hereunder:
The function named select_prefix, which is a member function of suffix tree data structure, is applied to find the longest common prefix between a given input string and a suffix in the tree. A prefix is simply a subsequence that begins at the beginning of the sequence and extends to any position of the sequence.
When we search common substrings between a string and a suffix tree, the select_prefix function is used to find the longest common prefix, then the common prefix is skipped and process reuses select_prefix to find another common prefix. This would be repeated until all characters in S2 are scanned. Thus, in a single scan of string S2, all common substrings can be identified.
In Figure 2, S1 has nine suffixes and they are all stored in its suffix tree. The square nodes are leaf nodes and represent suffixes. They are labeled by the start position of a suffix. The inner nodes are labeled by the length of the prefix of a suffix.
The process of picking out common substrings is shown step by step. At first, the string is “agcggcat,” S2 itself. The process traverses S1's suffix tree until mismatch occurs at the fourth character “g.” Then “agcg” is skipped and the string is updated to “ggcat,” the process traverses the tree for the second time until mismatch occurs at the seventh character “a.” Then “ggc” is skipped and the string is updated to “at.” The process repeats again and again until all characters in S2 are scanned. The selected common substrings are shown. “a” and “t” are omitted, although they are also selected.
3. The process of picking out common strings in S1 and S2 with sequence S2 and tree-S1 is run to form two common substring sets. The next step is dropping out ineligible substrings. Our standard for what constitutes eligible common substrings is described hereunder.
The criterion is that the starting positions of two matching substrings cannot be too far away. A pair of matching substrings is kept if the difference between their start positions is less than their length, otherwise they will be discarded. For highly similar sequences, most part of the sequences are the same. The matching of remote substrings would enlarge the areas that need to be aligned by dynamic programming algorithm so that the time of alignment is extended.
4. Next, the process aligns the remaining unmatched substrings between S1 and S2. Finally, all substrings are concatenated to form the aligned sequences.
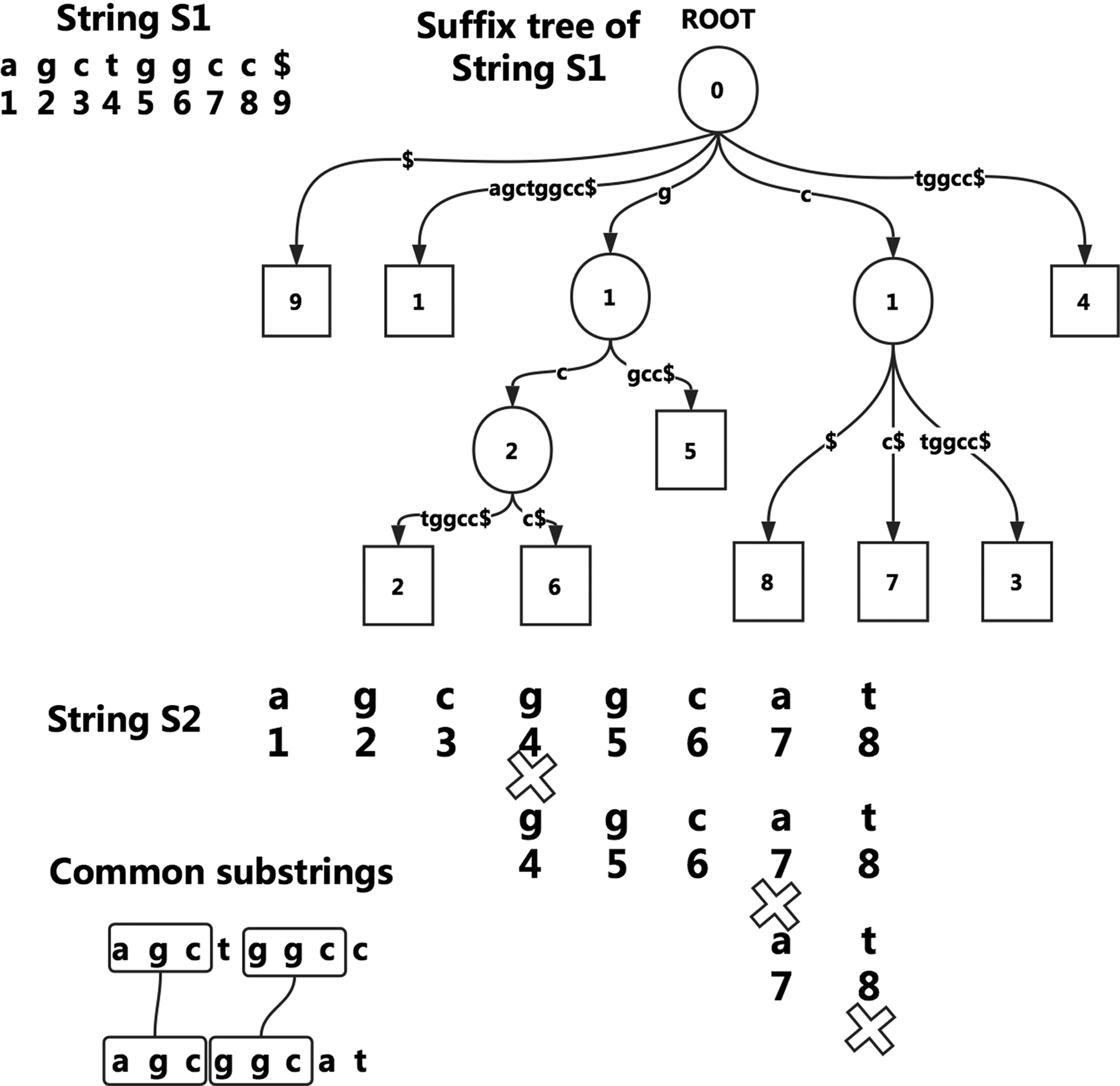
Suffix tree of a string and the example of selection of common substrings between “agctggcc” and “agcggcat.”
The construction of the suffix tree can be accomplished in O(n) time by Ukkonen algorithm, where n is the length of the sequence. Picking all common substrings can be done in a single scan of string S2, which can be accomplished in O(n) time where n is the length of S2. The alignments among unmatched substrings can be finished in O(
3.2. Center-star strategy
The center-star strategy (Zou et al., 2009) is a method that can solve the MSA problem with extremely high performance. And, the strategy's performance improves with increasingly similar sequences. For a set of sequences
The formula would help find out the sequence that is similar to all others. Here
After that, pairwise alignments are carried out between the center and the other sequences, one by one; each pairwise alignment gives two consequent sequences, the results of pairwise alignment. All the inserted spaces on center sequence are summed to obtain an aligned center sequence. At last, pairwise alignments between aligned center sequence and every sequence in S are carried out. The center-star strategy is shown in the following algorithm outline:
Input: sequence set
Output: aligned sequence set
1. Run pairwise alignment between every two sequences, and construct similarity matrix W, which is a symmetric matrix that stores the similarity scores of each of the two sequences in set S.
2. Select
3. Run pairwise alignments between the center and the other sequences.
4. Sum all the inserted spaces on center sequences to get the aligned center named.
5. Run pairwise alignment between aligned center and the other sequences again to get the result of the MSA.
6. Output the result.
When performing MSA among a set of similar sequences, it takes O(n) to finish a pairwise alignment with our suffix tree method previously illustrated. The construction of similarity matrix W can be accomplished in O(
3.3. Distributed and parallel implementation with Spark
As MASC can perform MSA in linear time with highly similar sequences, it has enormous potential for very high-throughput cases. However, in reality an ordinary computer cannot keep so many long sequences in memory, possibly leading to a memory crash, or wasting excessive time shifting between physical and virtual memory. Consequentially, we need to solve the memory storage problem, and parallelize our method using distributed parallel frameworks.
There are many frameworks available for handling large-scale data. MapReduce (Dean and Ghemawat, 2004) and Spark (Zaharia et al., 2010) are the most powerful and popular distributed computing frameworks. Spark is more suitable for this work, due to its memory computation characteristics.
The serial version of our method is illustrated in Figure 3.
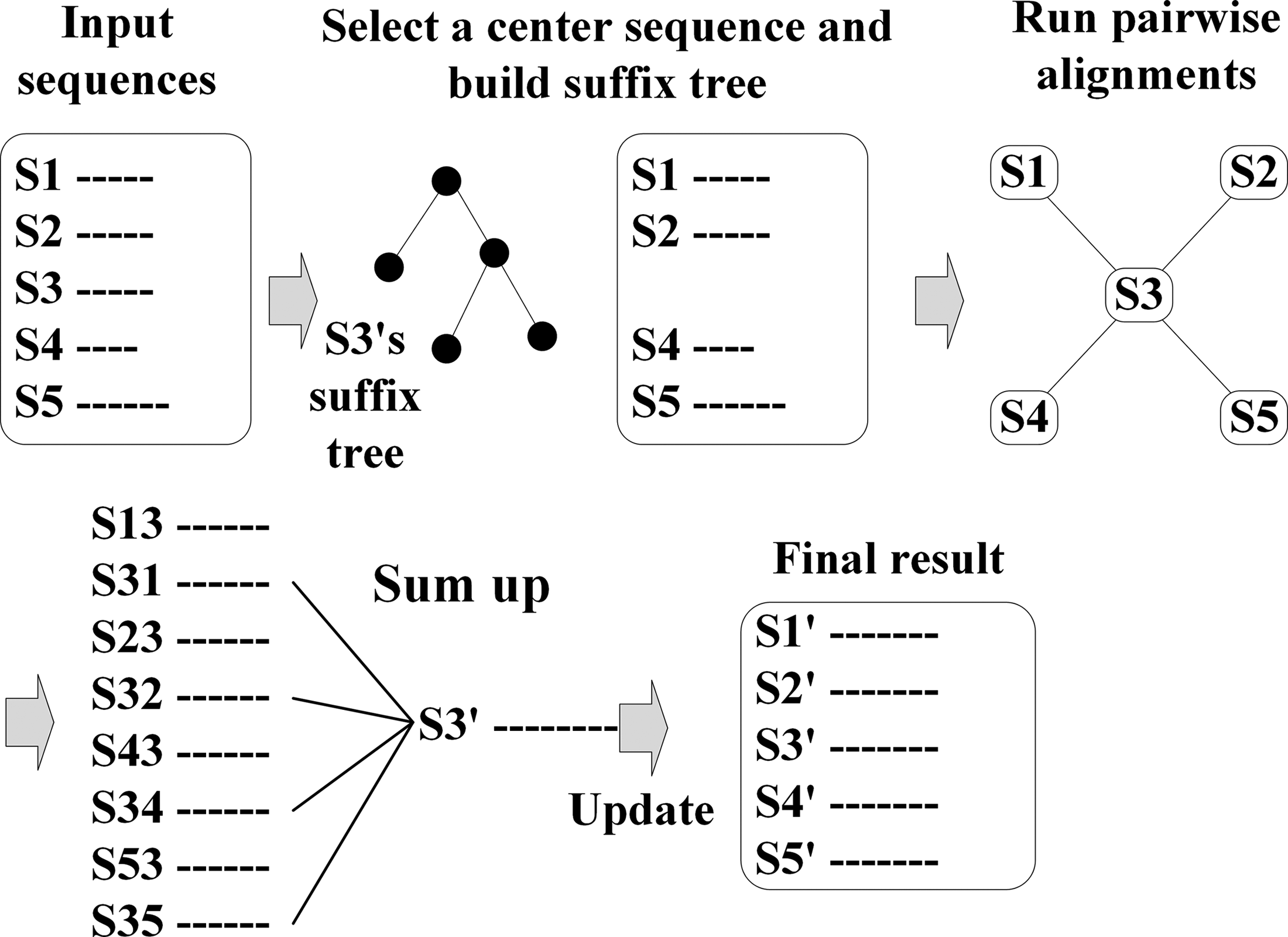
MASC flowchart. MASC, multiple sequence alignment based on a suffix tree and center-star strategy.
After inputting the data, the first task is to select a sequence as center and build its suffix tree. Then, the pairwise alignments between the center and all other sequences are run. Next, the process of summing up the spaces among sequences is carried out. Finally, the result is output.
We analyze the process to find “hot spots” within the program, the process of “pairwise alignment and sum up spaces,” which occupies most of the computational time, as shown in Figure 4.
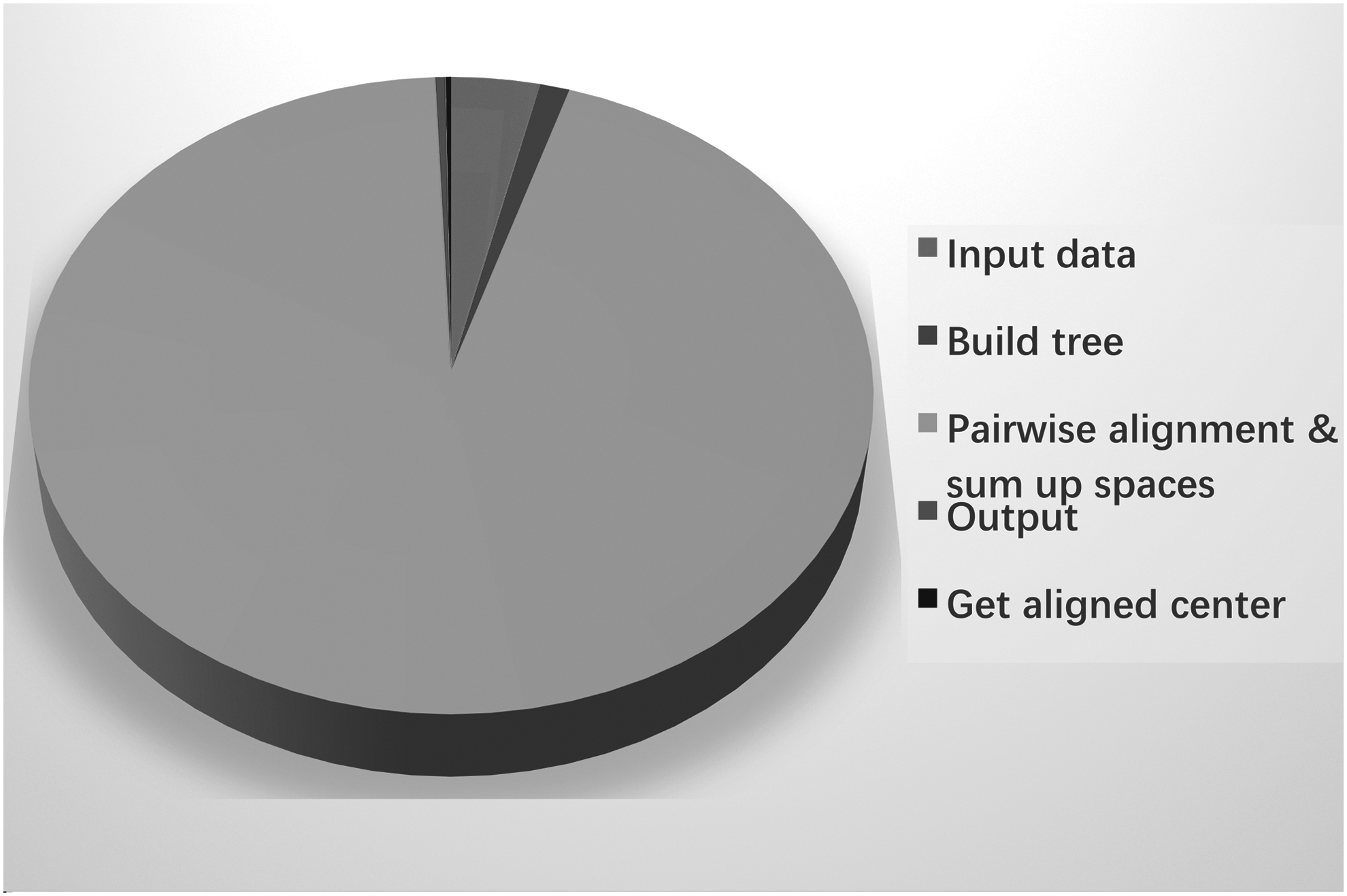
Time cost of MASC steps.
Fortunately, in MASC the process of alignment between the center and the other sequences can be parallelized, because there isn't too much data dependency.
According to Amdahl's law (Moncrieff et al., 1996):
where T is the total time that a program consumes, which consists of Ts and Tp. Tp is the time consumed by the part of the program that can be parallelized. Ts is the time consumed by the part of the program that must be run in serial. And p is the number of processors. So the speedup of our method is:
The limitation of the speedup of our program is 16 in theory. MASC was, therefore, developed within the Spark framework, and the process is shown in Supplementary Method. Supplementary Figure S1 shows details of the process of MASC. Supplementary Figure S2 shows the transformation of MASC's RDD in Spark. The Spark version of MASC is more than eight times faster than the original MASC in practice. The result of parallelization is shown in Section 4.5.
4. Experiments
4.1. Data sets and measurements
Balibase (Thompson et al., 2005) is regarded as the golden benchmark for most MSA research. However, the database is relatively small, and is only suitable for protein sequence alignment. Because there is no benchmark data set for addressing the large-scale nucleotide MSA problem, we employ human mitochondrial genomes (mt genomes) and 16S rRNAs as the test data in our experiment. Human mitochondrion genome is associated with Alzheimer's disease, Parkinson's disease, and type 2 diabetes (Tanaka et al., 2004). MASC may help analyze the function of mt genome. Our human mt genome data set contains 672 highly similar mt genome sequences, with a maximal length of 16,579 bp, and a minimal length of 16,556 bp. With the aim of testing the performance of our program with large-scale data, we duplicate the mt genomes 20 times, 50 times, and 100 times to enlarge the test set (Table 1).
Our main purpose of this project is to accelerate the MSA process and to improve the capacity to handle massive data. Therefore, we focus our attention on running time and throughput. We choose the sum-of-pairs (SP) value (Zou et al., 2009), which is a measurement of the quality of an MSA, for measuring alignment performance. The SP value is an integer representing the sum of every pairwise-alignment score from an MSA. For our purposes, in a pairwise nucleotide sequence alignment, if two nucleotides from the same column are different, one is added to the SP value, while if a space is inserted, two is added to the score, otherwise, if the two nucleotides are the same, the score remains unchanged. Thus, the SP value will be a positive integer, and a lower SP value indicates better quality of MSA. However, SP values are not suited for massive MSAs because the score may become too large and exceeds the computer's limitations. Therefore, we employ the average SP value instead, which is the SP value divided by the number of sequences (Zou et al., 2015).
4.2. Center sequence selection
This section is intended to prove that when we align highly similar sequences, it is feasible to select a center sequence randomly. A data set containing 600 homologous sequences selected randomly from mt genome data set is used to execute center-star MSA 600 times, which uses every sequence as center. Then the results of these experiments are quantitatively assessed by SP score. Then SP score is normalized and transformed into z-score. The formula is as follows:
Given,
Then,
We collect 600 z-scores and analyze the distribution of these data. The distribution is shown in Figure 5.
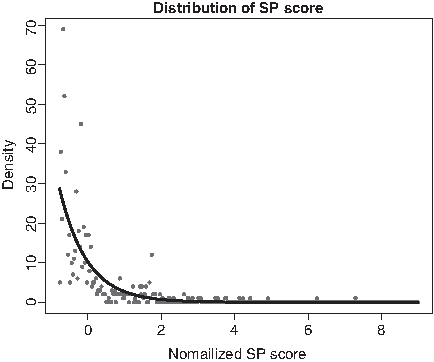
The distribution of normalized SP score. SP, sum-of-pairs.
From the fitting model, the probability that z-score is less than x is:
When an MSA is carried out with a randomly selected center sequence, the probability of z-score <4, which means that the SP score is <17,063, is 0.9983. The SP of the result of MAFFT and KAlign is between 16,500 and 17,000. That means random selection of center sequence is feasible.
4.3. Sequence similarity analysis
MASC is a method to do MSA among highly similar sequences. To find out which kind of sequences are suitable to be aligned by MASC, an experiment is designed to quantify the similarity of sequences. A 1000 bp long sequence is taken as basic sequence in the experiment. Then variations are carried on the copies of basic sequence. The data set is formed by basic sequence and variated copies. The variation is to change one residue of basic sequence to one element of {A, G, C, T, -}, where “-” means to delete the residue. The quotient of the number of residues that are changed divided by length of basic sequence is called mutation rate. In the experiment, mutation rate ranges from 0% to 100%. The result shows that MASC works when the mutation rate is no more that 54%.
In Figure 6, less SP value is on behalf of better accuracy. Before mutation rate gets to 33%, MASC has a better accuracy than KAlign, and the SP score and mutation rate are linearly related. When the mutation rate is >33%, KAlign has the best accuracy. MASC is more accurate than MAFFT when the mutation rate is no more than 54%. Because the variation is selected from {A, G, C, T, -} in random, five elements have the same probability to be chosen, so that the residue has 20% probability to be unchanged (e.g., when mutating a residue “A,” it has 20% probability to choose “A”). It means that the MASC works when there are >56.8% ([100% −54%] + 54%/5 = 56.8%) residues that are same between sequences that need to be aligned.

SP value of MASC, MAFFT, and KAlign with mutation rate changed.
4.4. Comparison with state-of-art tools
Most of the available state-of-the-art MSA software tools cannot address large-scale data. Therefore, we only perform comparisons with MAFFT and KAlign. KAlign and MAFFT are run on single nodes, without any parallel operations. Therefore, for fairness, we carry out all the experiments on the same server (Intel® Xeon® CPUE7-8890v3 @ 2.5 GHz with 2 TB total memory, and the Red Hat Enterprise Linux Server release 7.1 operating system). Table 2 shows time consumption for the various human mitochondrial genome data sets.
MASC, multiple sequence alignment based on a suffix tree and center-star strategy.
MSA is run with three software on different data sets that contains different number of sequences. From Table 2 we can see that MAFFT and KAlign take an extremely long time to finish the MSA among long sequences, even with relatively small files. Furthermore, KAlign cannot even handle data sets that have more than 13,440 sequences. However, MASC (serial version) is compatible with massive files of long sequences, and the parallelized version runs extremely faster than all other programs. The parallelization analysis is given in Section 4.5.
Table 3 shows a comparison of the average SP values among the different programs. As we have described previously, a lower SP value means better accuracy. Because the human mitochondrial genome data set sequences are highly similar, the different programs perform similarly. However, the data in Table 3 show that our method is somewhat more accurate than the other two methods with this data set.
MASC can be used for any file, no matter how large it is. In comparison, MAFFT cannot be used for files larger than 1 GB (67,200 sequences), and KAlign cannot be used for files larger than 10 MB (672 sequences). MASC is clearly superior for processing massive MSAs.
4.5. Speedup of Spark
We demonstrate the speedup and scalability of the Spark version of MASC in this section. The method we use is introduced in Supplementary Method. We perform our experiments on a server with 16 CPUs and 2 TB of memory (same specifications as previous).
We test the 1 × , 20 × , 50 × , and 100 × human mt genome data sets with different modes that run various number of executors. Figure 7 shows the results of those experiments. Each combination of number of executors (Zaharia et al., 2010) and data sets was run for 50 times. Here executors mean threads that do pairwise alignments.
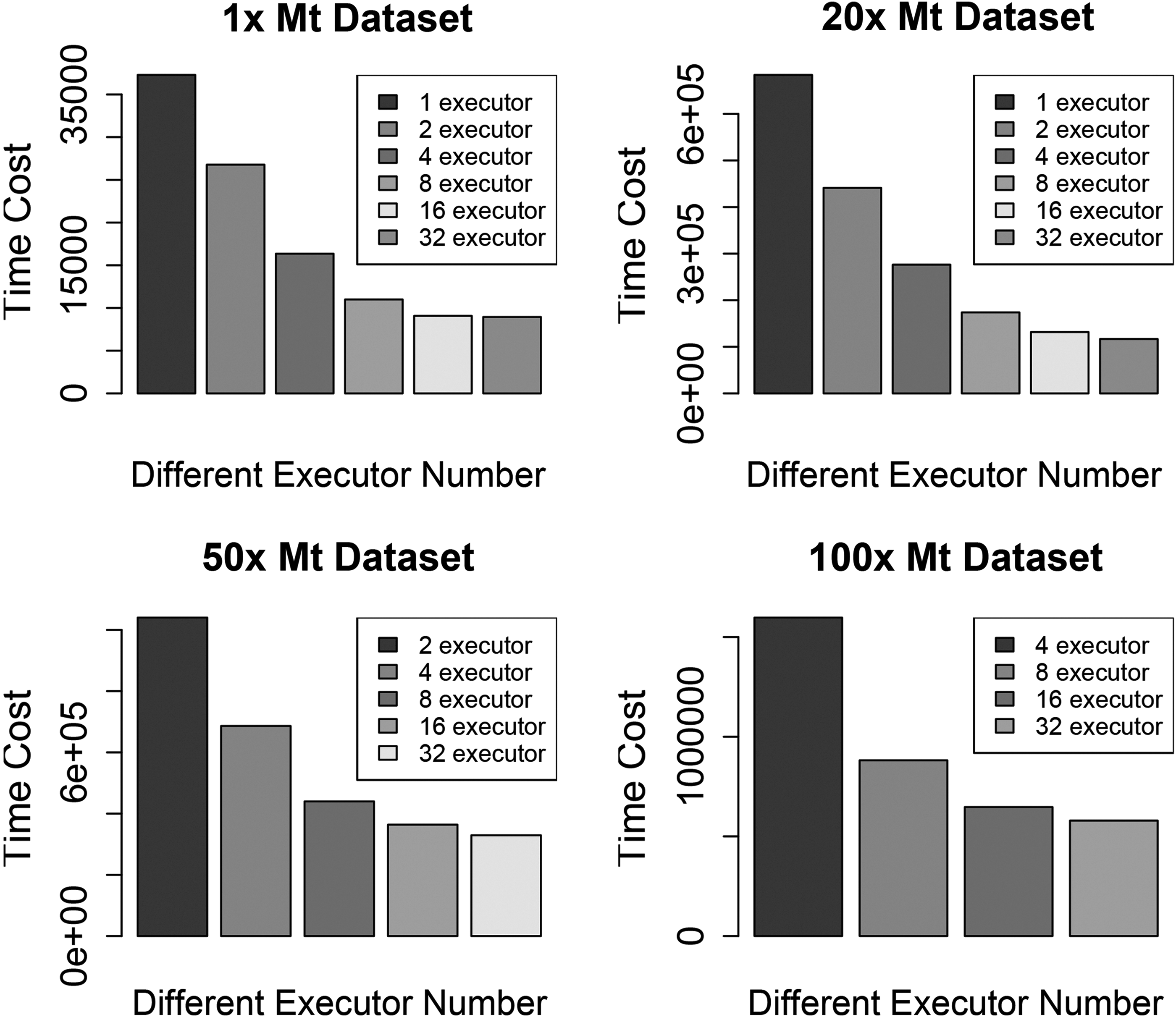
Running times for different mt genome data sets with different modes.
In Figure 7, each bar plot shows the performance of MASC on different data sets. Each column shows the average time cost of the program running in different environments (which can be regarded as different executors on the cluster). The unit of y-axis is second. We do not test the one- and two-executor modes for the 100 × data set, and the one-executor mode for the 50 × data set, because it is too difficult for one node keeping such a huge copy of massive data in memory. We see a remarkable speedup by Spark parallelization when we use no more than 32 executors at the same time. The speedup can be >10 sometimes, with an average total value of 8. The scalability is well maintained, and efficiency remains constant, while we enlarge the data set and the number of executors at the same time.
5. Conclusion
MSA is one of the most widely used modeling methods in biology, which is the basis of many analyses. These analyses include domain analysis, phylogenetic reconstruction, motif finding, etc. Recently the development of aligner includes the need of upscaling under the high-throughput sequencing pressure and the need for more complex sequence descriptors including noncoding RNA or nontranscribed genomic sequences. Likewise, the explosion of available genomic data has put a lot of pressure on the development of new aligners. Nowadays, parallelization is regarded as an important strategy to accelerate the process of MSA (Chatzou et al., 2016).
We propose MASC in this study, which can perform MSA in O(mn) time among highly similar sequences, where m is the number of sequences in the data set, and n is the average length of the sequences. MASC has high accuracy and performance. The core idea of our method is to accelerate the MSA process using three steps.
We speed up the process of pairwise alignment based on suffix tree, which is a powerful data structure for handling strings. Time complexity is O(n) at the most, when aligning pairs of similar sequences based on suffix tree. A center-star strategy is then employed as a heuristic to reduce MSA to pairwise alignments. MASC can be accomplished in O(mn) time when the sequences are highly similar and there is little loss in accuracy. Along with MASC's extremely high performance, we used the distributed parallel computing framework Spark to enlarge the memory of the system, and, in this way, the throughput of our program is substantially increased.
Extensive experiments with MASC were then performed. First, the accuracy and performance of our method were tested compared with other state-of-art tools. The scope of the comparison was quite limited, because most available tools are not optimized for performance nor efficiency. MAFFT and KAlign are two optimized tools available that were selected for comparison. The results of our experiments show that we have made some progress, and our method has better accuracy than the other two methods with our sample data sets, as indicated by lower average SP values.
MASC has been implemented on Spark and HDFS (Hadoop distributed file system) to handle the increasingly expansive data in the field of biology and bioinformatics. The method is very suitable for parallelization because the pairwise alignments between sequences are independent. In practice, the Spark version of MASC has great speedup and scalability. Both the single thread and the parallel tools are developed using Java, which works on multiple operating systems. Java 1.8 and Spark 2.0 are prerequisites for its operation. The codes and tools are accessible free of charge at (https://github.com/suwenhecn/MASC).
Although MASC has a good performance and accuracy, it still has some unsolved disadvantage. The ideal scenario for our method is aligning data sets without complex variation. The performance of MASC decreases a lot when it has to handle complex variations. As the sequences are not similar, suffix tree pairwise alignment gets less efficient because there are fewer common substrings and more large areas of unmatched pairs need to be aligned by Needleman–Wunsch algorithm. Apart from this, our improved center-star strategy could lead to an inaccurate result because it selects a center sequence randomly. For these reasons, we intend to develop MASC to adapt complex variations in our future work. Although the scope of application of MASC is limited in theory, it is still quite useful in many researches. To solve these tough problems, an algorithm with better robustness needs to be developed. MASC needs further development for more general applications.
Footnotes
Acknowledgments
This work was supported by National Key R&D Program of China 2017YFB0202600, 2016YFC1302500, 2016YFB0200400, and 2017YFB0202104; NSFC Grants 61772543, U1435222, 61625202, and 61272056; and Guangdong Provincial Department of Science and Technology under grant no. 2016B090918122.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.