
Abstract
Abstract
DNA repeats have great importance for biological research and a large number of tools for determining repeats have been developed. Herein we define a method for extracting a statistically significant subset of a determined set of repeats. Our aim was to identify a subset of repeats in the input sequences that are not expected to occur with a number of their appearances in a random sequence of the same length. It is expected that results obtained in such manner would reduce the quantity of processed material and could thereby represent a more important biological signal. With DNA, RNA, and protein sequences serving as input material, we also examined the possibility of statistical filtering of repeats in sequences over an arbitrary alphabet. A new method for selecting statistically significant repeats from a set of determined repeats has been defined. The proposed method was tested on a large number of randomly generated sequences. The application of the method on biological sequences revealed that for some viruses, shorter repeats are more statistically significant than longer ones because of their frequent appearance, whereas for bacteria, the majority of identified repeats are statistically significant.
1. Introduction
N
In NAs, there are three types of repeats related to their position in the sequence that have been investigated the most: (1) terminal repeats (e.g., long terminal repeats in viral genomes), (2) tandem repeats (e.g., satellite and mini- and microsatellite DNAs), and (3) interspersed repeats (e.g., transposable elements, SINEs and LINEs). Special types of short palindromic NA repeats are involved, for example, in the diversity of T cell receptor genes and are recognized as methylation or restriction enzyme-binding sites (Heringa, 1998). Repeats are also frequently found in proteins, although their role is less well understood (e.g., palindromic repeats are related to high α helical propensity in proteins, as well as to formation of protein–protein complexes) (Andrade et al., 2001).
The most investigated type of repeats is direct repeats, while other types have been more or less neglected. The majority of applications have been developed for finding repeats in NAs and less often in proteins. Also, a very few of the developed applications take into account the statistical significance of the obtained results. We present a novel method for the detection of statistically significant repeats in NAs (whole genomes) and in proteins (proteomes), aimed at improving the usability of the obtained results. We have also developed an application to implement the proposed method, available free for download (see the first Reference: StatRepeats).
1.1. Definition of repeats
In an arbitrary string, a pair of (sub)strings that satisfy certain conditions are called a repeat. Depending on the conditions they satisfy, repeats can be direct or inverse, which are further divided into complementary or noncomplementary subsets. In general, all variants of repeats can be precisely defined as follows:
Let A = {a,b,c,d,…} denote an alphabet with arbitrary symbols and L = {l1,l2,…,ln} is a language over alphabet A, which includes strings over A with an arbitrary length, including empty string, and let
Let the following functions be defined as:
• •
then, for all strings s∈L, the following four types of repeats can be defined (Fig. 1):
1. The substring pair
2. The substring pair (a,s,pa) and (b,s,pb) is an inverse noncomplementary repeat (IN) if and only if a = g(b) ∧ pa ≤ pb
3. The substring pair (a,s,pa) and (b,s,pb) is a direct complementary repeat (DC) if and only if a = f (b) ∧ pa < pb
4. The substring pair (a,s,pa) and (b,s,pb) is an inverse complementary repeat (IC) if and only if a = f (g(b)) = g(f (b)) ∧ pa ≤ pb
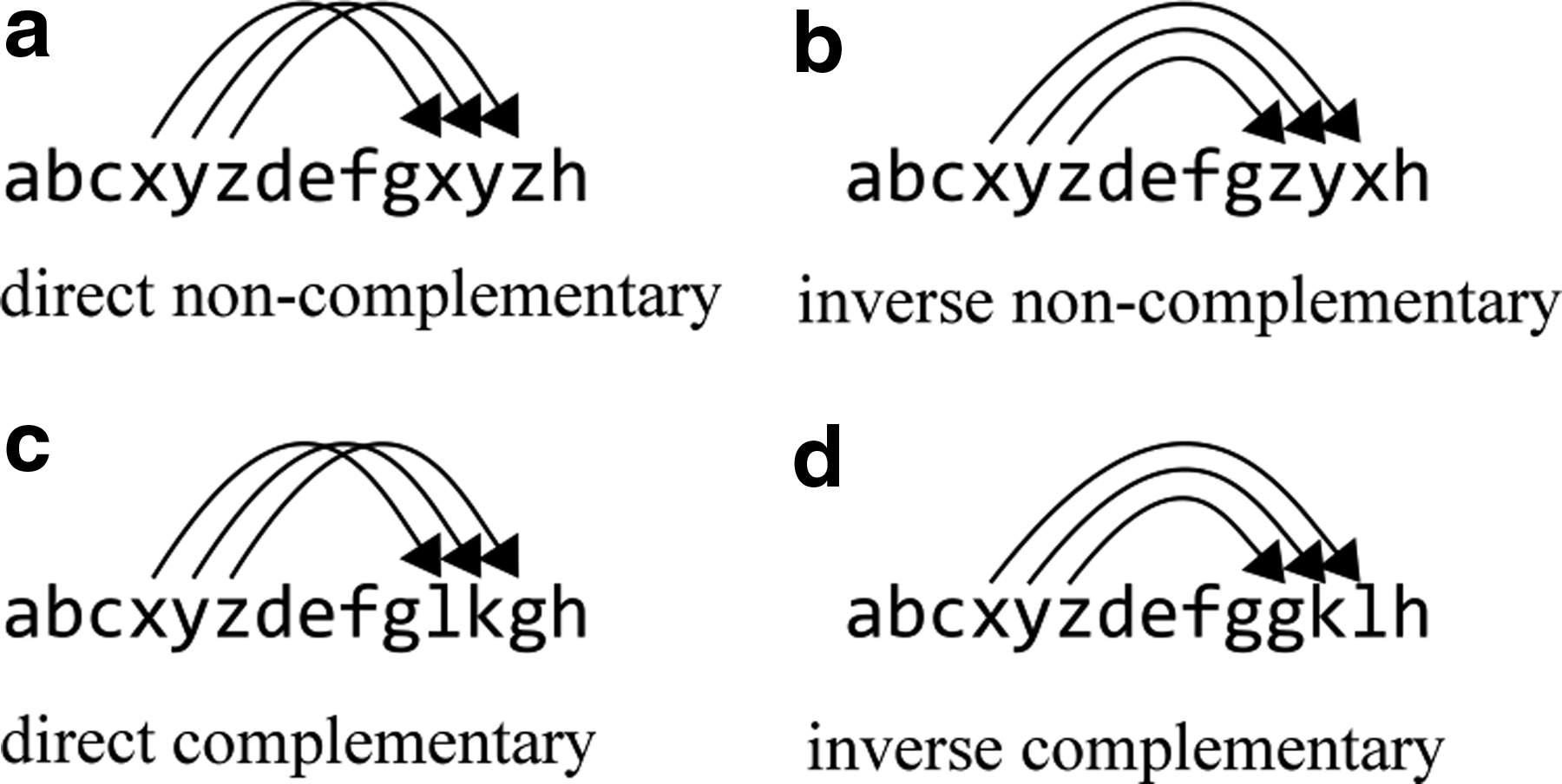
Graphical presentation of repeat types:
In the literature, different and often ambiguous terms for previously defined repeat types are often used, such as palindromes (for both types of inverse repeats), inverted repeats, everted repeats, and mirrored repeats (Gurusaran et al., 2013). Because of the manner in which various types of repeats were defined, the following synonyms can be used: mathematical repeats and mathematical palindromes for direct noncomplementary and inverse noncomplementary repeats in the case of these types of repeats in mathematics, and biological repeats and biological palindromes for direct complementary and inverse complementary repeats, with regard to the complementarity of their positions in sequences.
It is evident that if the substring pair (a,s,pa) and (b,s,pb) is a repeat, and if the elements of the pair include substrings a′ and b′, respectively, such that a = xa′y ∧ b = zb′w, then the substring pair (a′,s,pa′) and (b′,s,pb′) is a repeat of the same type. This is the reason why a maximal repeat is introduced. The definition of a maximal repeat is as follows:
1. The substring pair (a,s,pa) and (b,s,pb) is a maximal direct repeat if none of the substring pairs (xa,s,pa − 1),(yb,s,pb − 1) and (az,s,pa),(bw,s,pb) is direct repeats.
2. The substring pair (a,s,pa) and (b,s,pb) is a maximal inverse repeat if none of substring pairs (xa,s,pa − 1),(by,s,pb) and (az,s,pa), (wb,s,pb − 1) is inverse repeats.
Where x,y,z,w∈A. For brevity, from this point on the repeat we are referring to is a maximal repeat.
Although these definitions are based on linear string only, previous general definitions cover all cases of repeat occurrences in biological materials. Elements of substring pair in such repeats can be separated by gap (with a specified length), or can overlap. Also, given definitions are valid for an arbitrary input alphabet, which permits exploring DNA sequences (A = {A, C, G, T}), RNA sequences (A = {A, C, G, U}), protein sequences
(A = {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y, U, O}), as well as work with another user-defined alphabet. Given the important role of repeats in the analysis of biological material, there is a clear need to determine which repeats found in the input material are significant with regard to the content of the material itself. Our primary assumption is that if a group of repeats occur with a high probability in a random sequence, then that group is most likely to represent a random pattern, and that the existence of such a pattern is not significant for the organization of a biological process.
1.2. Motivation
The process of finding repeats, especially in large input material, produces a mixture of repeats with a statistically expected occurrence and repeats with a lower probability of occurrence in the input sequence. The separation of statistically important results allows us to obtain a clearer outcome, to separate an important signal(s) from noise. The aim of the research was to define the method which, based on the specified p-value, would allow for filtering of the set of repeats found and selection of only statistically significant repeats. In practice, extracting repeats from biological material is often the first step in research. The number of repeats found can be huge. By applying the proposed method, the amount of material for further processing is reduced, while data likely to represent an important biological signal are retained and data that probably occur in a random sequence of the same length are rejected.
1.3. Related work
Repeat finding algorithms can be divided into two groups: those that use an annotated library of known repeats and the second group of de novo repeat finders. An example of the first type is RepeatMasker (Smit et al., 2013–2015) where a repeat is defined as a substring that occurs frequently in input sequence and uses a library of previously known repeats. Methods from the de novo group find repeats without prior knowledge of found repeats using only the input sequence (Martinez, 1983). Among these methods, some use heuristics such as Repex (Gurusaran et al., 2013) and Emboss (Rice et al., 2000), while others detect repeats precisely like REPuter (Kurtz et al., 2001). A lot of methods for finding tandem repeats are available (Benson, 1999; Wexler et al., 2005; and Landau et al., 2001). A number of algorithms have been developed to identify repeats in proteins, such as GBA (Li and Kahveci, 2006).
We focus on de novo methods precisely finding repeats. REPuter (Kurtz et al., 2001) provides statistical significance of repeats by computing the number of repeats of the same length or longer that one would expect to find in a random DNA of the same length as the input sequence. Repseek (Achaz et al., 2007) calculates the smallest length above which no such repeat is expected to occur by chance in a random sequence and uses it as a seed or uses the score of the best local alignment observed between two random sequences (Waterman and Vingron, 1994), which is also used in Swelfe (Abraham et al., 2008). Finding the minimal length above which repeats are significant and avoid spurious matches has been studied (Guyon and Gunoche, 2008), and furthermore, finding the optimal minimal length (Devillers and Schbath, 2012) is also recoverability of sequence from words (Arratia et al., 1996). We developed an easily computable (Robin and Schbath, 2001) method that considers only exact maximal repeats in an input sequence as a basic repeat type and filters them based on a defined p-value.
The main difference from previously mentioned statistical methods is that our method does not group repeats by their length only, as within that length some repeats may indeed occur many times thus making them statistically significant, but others may occur just a few times. Instead of the coarse measure of significance based on the repeat length, we look at the number of occurrences of each individual repeat and calculate the probability that the specific number of occurrences of that specific repeat of specific length being significant.
2. Methods
Our method defines the procedure for detecting whether the occurrences of concrete repeats in an input string are statistically significant. We therefore calculated the expected number of lexicographically different repeats (maximal pairs) that occur kl times in a random sequence using statistical distributions. The significance of a given repeat occurring kl times was determined by comparing the calculated expected number with the actual number of repeats found. The correctness of the calculated results was checked by comparing them with the counted number of repeats of the same length occurring kl times in randomly generated sequences with the same properties (length of input sequence N over an alphabet with cardinality c).
2.1. Probability for an arbitrary number of substring occurrences
If every symbol in an alphabet has the same probability of occurrence, then the expected number of occurrences of a substring with a length l in string s with a length N using an alphabet with cardinality c is
The probability that an arbitrary substring occurs k times can be approximated using the binomial or Poisson distribution, even though the events of a substring occurring at different positions in a string are not independent:
The previous formula is suitable if substrings do not overlap with other identical substrings. Otherwise, they have a tendency to accumulate (to appear in droves), in which instance Formula (2) does not provide an adequate approximation. In such cases, the compound Poisson distribution (Robin and Schbath, 2001) can be used:
where p is the probability of an overlapping pipe occurrence.
For a small value of λ, previously specified distributions are used. However, for a larger λ, that is, when a substring occurs frequently, a version of the normal distribution can be used instead. If substrings are not self-overlapping, then the following distribution can be used (Ewens and Grant, 2005):
and if substrings can be self-overlapping then the distribution
can be used, where ξj = 1 if the first j symbols are equal with the last j symbols of the same substring, otherwise, ξj = 0.
Because substrings that are self-overlapping are rare (and as we examine groups of repeats), they can be ignored in calculations, and the probability for an arbitrary number of substring occurrences with fixed length is calculated with the Formulae (2) and (4).
2.2. Probability of an arbitrary number of repeat occurrences
The probability of occurrence of z substring pairs can be calculated using formulae for determining the probability of a string occurring k times. Multiplying this number with the probability that a certain number of these pairs are maximal provides the probability of an arbitrary number of repeat occurrences. As defined previously, a repeat is maximal if and only if it cannot be extended to a longer repeat. The probability for this is equal to the probability that the letters surrounding both corresponding substrings of a pair do not correspond to each other, which is
2.2.1. Direct noncomplementary repeats
In the case of direct noncomplementary repeats, the pair of substrings that represent this repeat consists of the same string in different positions in the input sequence. From n substrings,
where n is the minimal value that satisfies inequality
We could not find the formula for determining the values for
The limit for n in these calculations depends on the available computing resources and becomes very expensive as the alphabet cardinality gets higher. So to get results for larger n, we generated a large number of random sequences of 2n length and counted all possible combinations of symbols to the left and to the right of substrings that form a pair. We calculated values for n < 32. For larger n values, we found that the binomial distribution
2.2.2. Direct complementary repeats
In the case of complementary repeats, a pair that represents a repeat consists of mutually different substrings. The probability of occurrence of z such substring pairs can be calculated using the following:
where div(z) represents all divisors of z. If
where
2.2.3. Inverse noncomplementary repeats
In the case of inverse noncomplementary repeats, the pair of substrings that create the repeat are different strings, except in the case of strings that are palindromes themselves. From n substrings that are palindromes themselves,
where n is the minimal value that satisfies the inequality
where
2.2.4. Inverse complementary repeats
In the case of inverse complementary repeats, the pair of substrings that create the repeat are different strings, except in the case of strings that are palindromes themselves.
As only strings with even length can be palindromes themselves, we use the calculation for inverse noncomplementary repeats for repeats with even length and for repeats with odd length we use the calculation for direct complementary repeats.
2.3. Determining whether a repeat is statistically significant
The computed expected value E represents the mean of normal distribution, which models the number of different repeats of length l that occur kl times in a randomly generated sequence of length N over an alphabet with cardinality c. With the given p-value, we can then calculate the boundary above which the results are statistically significant.
2.4. Limitations of method
The described method has two limitations: it assumes that all letters in the alphabet have the same frequency of appearance and it buckets all repeats of length n occurring k times together even though overlapping repeats have a higher chance to accumulate.
Both limitations are a result of a search of a computationally feasible method for improving the signal-to-noise ratio of found repeats. Any signal-to-noise filter will have both false positives and false negatives, and we tried to find a balance between the quality of the filter and execution times.
The first limitation lies in our assumption that all base pairs have the same frequency in a random sequence. Modifying our method to work with nonidentically distributed base pairs (where formulae include weights) requires that for every distribution, appropriate probabilities must be computed whether the found repeats are maximal. This would make our method computationally unfeasible and prevents implementation of an efficient program.
The bucketing of all repeats by length and number of occurrences underestimates the number of overlapping repeats that occur in a random sequence, and leading to a slightly higher number of false positives. However, highly overlapping repeats of meaningful length are a tiny fraction of all repeats found, so increasing the computation time by several orders of magnitude to avoid a slight increase in false positives was evaluated to be a bad trade-off. Likewise, as long as the base pairs are uniformly distributed, applying a method that more precisely models overlapping repeats (such as Markov chains) would impose prohibitive performance penalty while providing a negligible improvement in precision (Robin and Schbath, 2001).
For these reasons, we rely on bucketing per repeat length and the number of occurrences to keep the computational cost down, as well as on computationally expensive precomputed probability tables to model how many repeats in a found group are maximal as those are not independent. Despite these limitations, our method produces a significant benefit in result quality over nonfiltered results.
3. Method Implementation
The proposed method is implemented in the program StatRepeats. The implementation has four phases: finding all maximal repeats in an input sequence, calculating the expected number of their occurrence, determining which of the found repeats are statistically significant, and outputting the statistically significant repeats. In the first step of the program, we found all maximal repeats in an input sequence of length N over an alphabet with cardinality c. This problem has been known for a long time and it was first solved using suffix trees (Gusfield, 1997), while subsequent solutions have used suffix arrays (Abouelhoda et al., 2004) as they are more compact in memory and are linear in both time and space (Ko and Aluru, 2003; Kim et al., 2003). At present, a considerable number of very efficient algorithms for suffix array construction are available. In StatRepeats, we used a slightly modified divsufsort algorithm (Mori, 2006).
Calculation of the expected number of repeats in the second step was performed as described in Section 2. For direct noncomplementary and inverse repeats, the values P(n,kl) were precomputed and built into the program for alphabets with cardinalities between 4 and 22 and with limits n < 32, while for larger values of n we used calculations described in the method definition. For direct (inverse) complementary repeats, we precomputed the values
In the third step, the program can perform or skip statistical filtering. If filtering is not chosen, the program can find all repeats starting from a certain length in any input sequence. If statistical filtering is chosen, the program provides very precise results for any type of repeat in the input sequences over alphabets with cardinality between 4 and 22, and good results for direct complementary repeats with alphabets of any cardinality. In the case of inverse complementary and noncomplementary repeats (direct and inverse) over alphabets whose cardinality is not between 4 and 22, statistical filtering is not provided in the current version of the program but will be added in the future. In case of working with amino acids, our program also provides mappings for different groups of amino acids such as aromatic, charged, and polar, leading to alphabets with cardinalities between 8 and 22. The program can output results in many different ways. Console output and file output (in a number of different formats) are used most often, and are well structured so that they can be easily utilized by another program. Alternatively, the program can output repeats to an ODBC-compliant database, thus enabling simplified Data Mining or other Big Data approaches.
StatRepeats is written in C++ and is available for Linux and Windows (StatRepeats). Mandatory input arguments are names of file with the input sequence and minimal repeat length that we are searching for. For other input options, the default values that can be changed are defined. A detailed explanation of program arguments and different types of results that can be produced with StatRepeats can be accessed on the website where the program is available. Comparison of the program characteristics with (some) similar programs is given in Table 1 (possibilities not connected to repeats are not considered).
ACGT, ACGT-Iupac base nucleotide codes; IUB, Iub-Iupac base wildcards: R,Y,M,K,W,S,B,D,H,V,N.
3.1. Method correctness verification
As we used a number of approximations in the method, it was important to measure the magnitude of error against a large number of data sets. Verification of the method was performed by comparing the results (computed by previously explained formulas) for expected values for a random sequence of length Ntest over an alphabet with cardinality ctest, with the average number of repeats found in 1000 or more sequences generated using the Mersenne Twister 19937 (Matsumoto and Nishimura, 1998) pseudorandom number generator, with same length Ntest over an alphabet with cardinality ctest. The previously described method proved accurate, with a low relative error. The error slightly increased as the expected values get closer to zero. The differences between the computed expected values, the found repeat counts, and the percentage differences between them are shown in Figure 2.
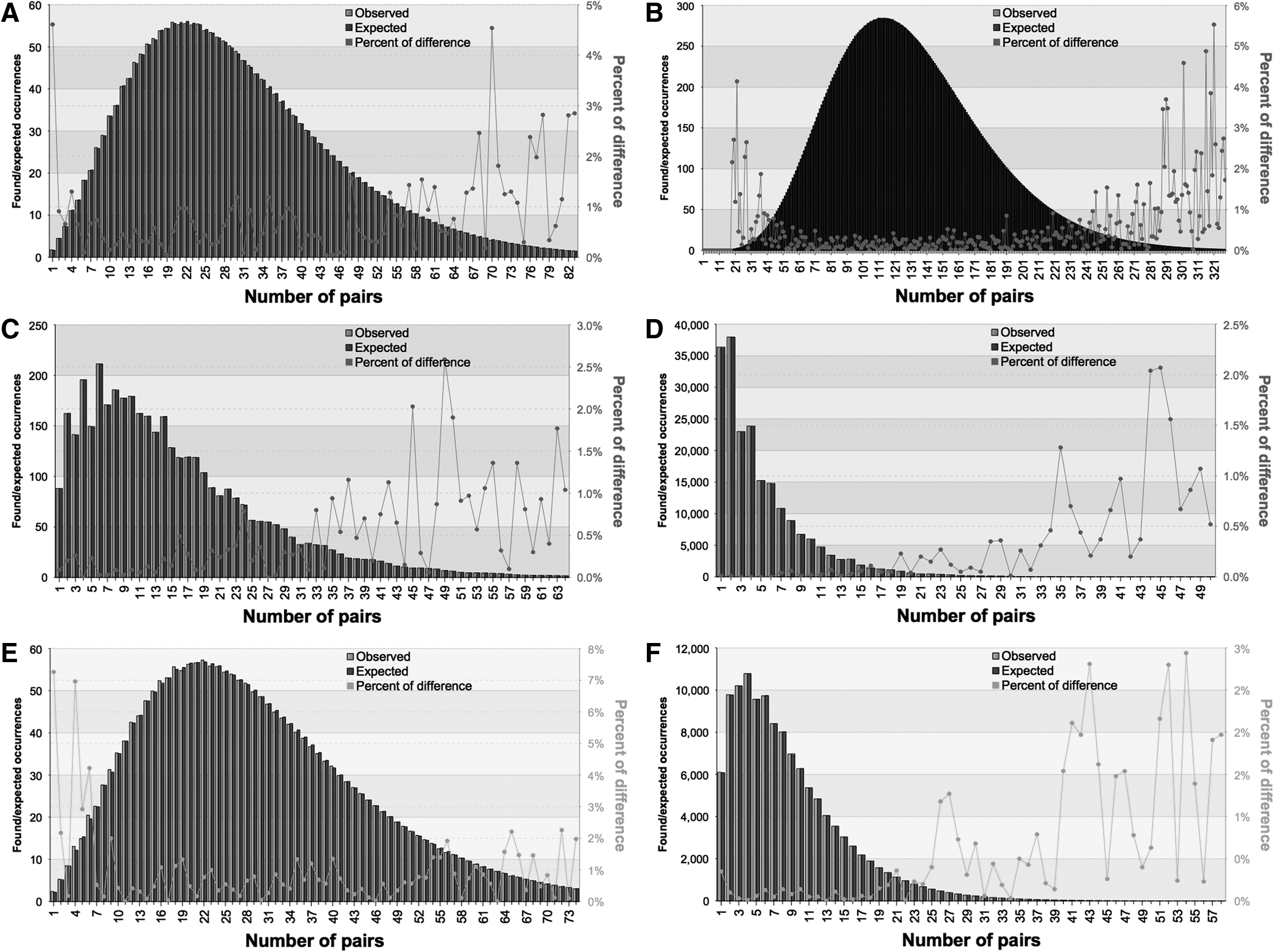
Differences between repeat counts in a random sequence and computed expected number. x-Axis represent the number of pairs (repeats); primary y-axis represents number of event (found or expected repeat pairs) occurrences; and secondary y-axis represents the percentage of differences between found and expected values. Values for DC, DN, and IC repeats with length 6 in a sequence of length 30,000 are shown on subfigures
As the repeat length that we are searching for becomes lower, there is a growing dispersion of results and the calculated expected values get very close to zero. All of the expected outcomes have very small probabilities, but in any concrete experiment, some of those outcomes will ensue. In these cases, all found results have counts greater than the expected value, which implies that all of the repeats found are statistically significant. Hence, the implemented program proposes a lower limit (reached empirically) for a repeat length under which filtering is not provided.
3.2. Examples of program application
The designed program has been tested on different sets of nucleotide and protein sequences. Sets of sequences and obtained results are available in the program package. The longest sequence on which the program was tested is human chromosome 21 GRCh38.p2 (NCBI ID: NC 000021.9) with 46,709,983 bp. In Table 2, we show a part of the results that were obtained by applying the program that searched for all four types of repeats with or without statistical filtering. We used the following genomes: Tobacco mosaic virus (NC 001367), Saccharomyces cerevisiae S288c chromosome I (NC_001133), Halophilic archaeon DL31 (NC 015954), Escherichia coli str. K-12 substr. MG1655 (NC_000913), as well as the following proteins: insulin receptor long proprotein isoform [Homo sapiens] (NP 000199), epidermal growth factor receptor, isoform a precursor [Homo sapiens] (NP_005219), and amelogenin X, isoform 1 precursor [Homo sapiens] (NP 001133).
The size of the material for which the program can be applied is limited by the size of the main memory of the computer. StatRepeats uses O(N) bytes, where N is the input sequence length. More precisely, 13N bytes of memory for finding all repeats in case of direct noncomplementary repeats, and for other repeat types, 16N bytes are needed. If statistical filtering is used, then for every unique repeat substring, an additional 12 bytes are needed in a 32-bit system, and 24 bytes in 64-bit systems. The execution time depends on the length of input sequence, minimal length from which repeats are searched for, number of found results, and the speed of the output device.
Applying StatRepeats with and without statistical filtering provided results that could be significant in further research. In many examples of previous research, the repeat length from which the repeats were searched for was determined arbitrarily relative to the input sequence length with the assumption that the longer the repeats were, the greater was their significance. However, the obtained results showed that in some genomes, shorter repeats are more statistically significant than longer ones. In Table 3a, we show the results for the murine hepatitis virus strain JHM genome where we searched for direct complementary repeats starting from length 5. Counts are shown for each length individually, with and without statistical filtering. The obtained results revealed that the longer repeats are less statistically significant than shorter repeats. In general, when starting from some repeat length, all found repeats were expected to be statistically significant; however, in this particular case, such long repeats are not present in the input sequence.
Figure 3 shows the results for the same genome for repeat length 6. It can be seen that for some number of pairs, the number of occurrences is much higher than the expected number that renders them statistically significant. Determining the significance of repeats based on minimal length only does not provide precise filtering.
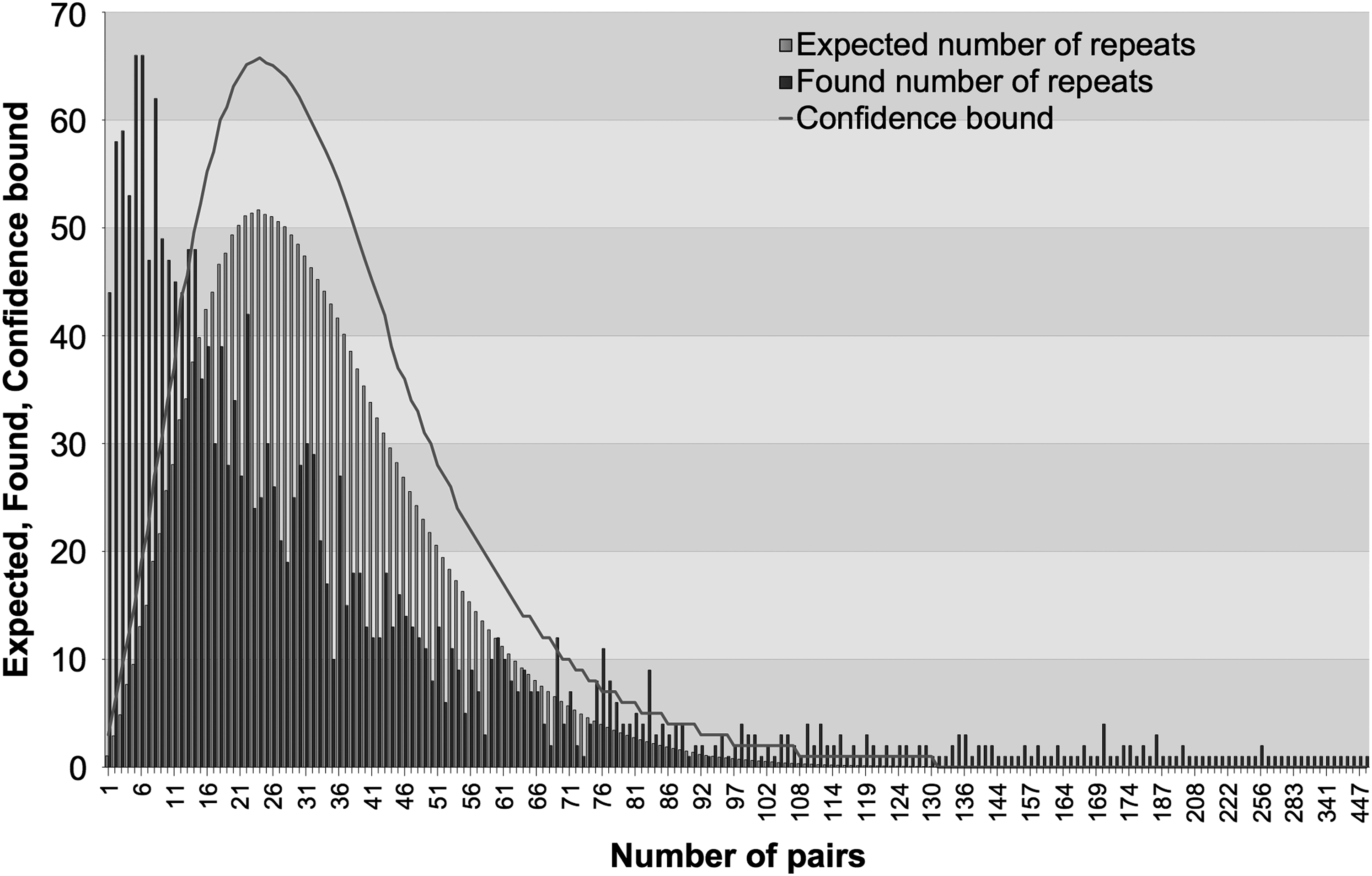
Statistically significant DC repeats of length 6 for murine hepatitis virus strain JHM (AC 000192). Confidence bound is the boundary above which the results are significant.
An example similar to the previous one was obtained for Enterobacteria phage Mu (Table 3b). In this example, all repeat lengths had low filtering survival. Once again, as the repeats got longer the filtering survival became lower. In the set of genomes we tested, this was not the prevailing trend. For the majority of genomes we applied our program to, the trend was that as the repeats get longer, the percentage of filtering survival rises. An example that illustrates this is Lactococcus phage bIL67, which we searched for direct noncomplementary repeats (Table 4).
4. Conclusion
We have developed a method for the precise determination of statistically significant repeats of all types (direct, inverse, complementary, and noncomplementary). The method can be applied to both nucleotide and amino acid sequences, as well as to sequences with a user-defined alphabet. The program that implements the method was tested on various nucleotide and amino acid sequences. It was noted that not all repeats found were statistically significant, and in some cases, counterintuitively, shorter repeats were statistically more significant then longer ones. For the large number of repeats that can be found in genomes, if the minimal repeat length is relatively low, our method provides a significant gain in performance and quality of results compared with methods that output all found repeats.
Footnotes
Acknowledgment
This work was partially financially supported by the Ministry of Education, Science and Technological Development, Republic of Serbia, Project No. 174021.
Author Disclosure Statement
No competing financial interests exist.