Constructing expression networks using transcriptomic data is an effective approach for studying gene regulation. A popular approach for constructing such a network is based on the Gaussian graphical model (GGM), in which an edge between a pair of genes indicates that the expression levels of these two genes are conditionally dependent, given the expression levels of all other genes. However, GGMs are not appropriate for non-Gaussian data, such as those generated in RNA-seq experiments. We propose a novel statistical framework that maximizes a penalized likelihood, in which the observed count data follow a Poisson log-normal distribution. To overcome the computational challenges, we use Laplace's method to approximate the likelihood and its gradients, and apply the alternating directions method of multipliers to find the penalized maximum likelihood estimates. The proposed method is evaluated and compared with GGMs using both simulated and real RNA-seq data. The proposed method shows improved performance in detecting edges that represent covarying pairs of genes, particularly for edges connecting low-abundant genes and edges around regulatory hubs.
1. Introduction
With the accumulation of whole-genome expression data, there is an increased interest in depicting patterns of expression variation at the transcriptome level. Genes exhibiting covarying expression levels may be regulated by common factors and participate in related functions; therefore, statistically constructed expression networks can generate candidate gene sets with shared regulatory mechanisms or shared biological function. Gaussian graphical models (GGMs) offer an elegant framework to investigate the covarying patterns of many genes (Li and Gui, 2006; Meinshausen and Bühlmann, 2006; Yuan and Lin, 2007; Peng et al., 2009). Under such models, gene expression data are represented as an \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n \times d$$
\end{document} matrix, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{X}} ,$$
\end{document} with each row representing a sample and each column representing a gene. A GGM assumes that the samples (rows) are independently and identically drawn from a Gaussian distribution with mean \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mu$$
\end{document} and covariance matrix \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$ \mathbf{\Sigma} $$
\end{document}, where the concentration matrix, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mathbf{\Sigma}^{- 1}$$
\end{document}, depicts an undirected graph, in which each node represents a gene, and two nodes are connected by an edge if and only if the corresponding entry in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mathbf{\Sigma}^{ - 1}$$
\end{document} is nonzero.
In many gene expression studies, the number of genes far exceeds that of the sample size \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( d > n ) ,$$
\end{document} and hence the maximum likelihood estimate of the concentration matrix is problematic because the sample covariance matrix is either singular (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$d > n$$
\end{document}) or has high variance \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( d \approx n )$$
\end{document}. Under a reasonable assumption that only a small fraction of genes are under coordinated expression after accounting for the expression levels of the remaining genes, a number of methods have been proposed to estimate a sparse concentration matrix. Meinshausen and Bühlmann (2006) proposed to fit a lasso regression for each gene; extending this approach, the space method jointly models all entries in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mathbf{\Sigma}^{ - 1}$$
\end{document} (Peng et al., 2009). In parallel, graphical lasso proposes to estimate \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\textbf{A} = {\mathbf{\Sigma} ^{ - 1}}$$
\end{document} by maximizing a penalized likelihood function (Yuan and Lin, 2007; Friedman et al., 2008). These approaches have been widely applied to microarray gene expression data to address a variety of questions (Oh and Deasy, 2014).
Measuring expression level by RNA sequencing has become a powerful tool for studying gene regulation in human and nonhuman genomes. However, modeling the covarying pattern using RNA-seq data poses a unique challenge because the measurements are non-negative counts and do not follow a Gaussian distribution. For certain purposes, univariate quantile transformation may be adequate for downstream analysis; however, such a transformation distorts the correlation structure among genes, especially when counts are low or have many ties.
A number of studies have developed high-dimensional graphical models for non-Gaussian data, considering binary (Ravikumar et al., 2010; Wang et al., 2011), multinomial (Jalali et al., 2011), or nonparametric models (Liu et al., 2009, 2012; Dobra et al., 2011). For count data, a series of approaches have been proposed, modeling the marginal distributions of the read counts as Poisson distributions (Allen and Liu, 2013; Yang et al., 2013). These studies show that it is difficult to construct a multivariate joint Poisson distribution that simultaneously permits global conditional independence, as well as both positive and negative conditional correlations. Current solutions to overcome these challenges either focus on the joint distribution in local neighborhoods or truncate the observed reads. Another limitation is that the RNA-seq counts often show overdispersion, so modeling them by Poisson distributions may not be appropriate (Robinson et al., 2010).
Here we propose a novel statistical model based on a Poisson log-normal (PLN) distribution, which offers an intuitive framework to model the conditional dependency of RNA-seq count data while allowing for overdispersion in the marginal distributions. In essence, this model assumes that the observed RNA-seq counts are Poisson variables with mean parameters determined by the underlying expression levels, where the logarithm of the underlying unobserved expression levels follows a multivariate normal distribution. Our goal is to estimate the GGM for the underlying expression levels. Similar to graphical lasso, PLN uses an \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \ell _1}$$
\end{document} penalized likelihood function to achieve sparsity of the estimated network. We use Laplace's method to approximate the likelihood, and implement an alternating directions method of multipliers (ADMM) algorithm (Boyd et al., 2011) to solve for the penalized maximum likelihood estimator (MLE). Simulation studies reveal that PLN performs favorably compared with GGM, when the latter is applied to RNA-seq data with or without various transformations. The improvement is particularly apparent for low-abundance transcripts and/or under weak signals. Furthermore, PLN achieves greater sensitivity in detecting highly connected genes (hubs), which point to master regulators. Finally, we apply PLN to a large RNA-seq data set (Lappalainen et al., 2013) in humans, and construct networks that generate hypotheses of genetic regulatory interactions.
2. Methods
2.1. PLN model
In this section, we describe the proposed PLN model for estimating a sparse undirected network using RNA-seq read count data. Let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{X}} = { \left[ {{{ \bf{x}}^1} , {{ \bf{x}}^2} , \ldots , {{ \bf{x}}^n}} \right] ^T}$$
\end{document} be an \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n \times d$$
\end{document} matrix representing the observed counts for n samples and d genes. We assume the n samples are independent, and the entries of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${{ \bf{x}}^i} = ( x_1^i , \cdots , x_d^i{ ) ^T}$$
\end{document} are conditionally independent, given the underlying unobserved expression levels of the ith sample whose logarithm is denoted by \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bm{\eta} ^i} = ( \eta _1^i , \cdots , \eta _d^i{ ) ^T}$$
\end{document}.
where Ki is a scaling factor adjusting for library size of the ith sample. Although the observed counts of mapped reads also depend on the length of the gene, it is equivalent and notationally more convenient to account for this factor in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\eta _j^i$$
\end{document}, because the length is constant for each gene across all samples. We further assume that the underlying expression levels follow a multivariate log-normal distribution, that is,
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{{\mathbf{\eta}}^i} \mathop \sim \limits^{{ \rm{i}}{ \rm{.i}}{
\rm{.d}}{ \rm{.}}} N ({\mathbf{\mu}} , {\mathbf{\Sigma}}) , i = 1
, \cdots , n.
\end{align*}
\end{document}
Our goal is to estimate the concentration matrix, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{A}};$$
\end{document} in particular, we aim to identify nonzero entries of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{A}} ,$$
\end{document} which indicate conditionally covarying pairs of genes. The Gaussian mean parameter, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\bm{\mu} ,$$
\end{document} is treated as a nuisance parameter. The scaling factors, Ki, are assumed to be known constants. To encourage sparsity, we seek a positive definite \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{A}}$$
\end{document} that minimizes an \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \ell _1}$$
\end{document} penalized log-likelihood function:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
O ( \bm { \mu } , { \bf { A } } ) = - \frac { 1 } { n } \mathop \sum \limits_ { i = 1 } ^n \log ( { L_i } ( \bm { \mu } , { \bf { A } } ) ) + \lambda \mathop \sum \limits_ { j \ne k } \; { W_ { jk } } \vert { A_ { jk } } \vert , \tag { 2 }
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${W_{jk}}$$
\end{document} is a weight that allows differential amounts of regularization of the entries of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{A}}$$
\end{document} based on prior knowledge or anticipated network properties (see Supplementary Data).
2.2. Computational algorithm
Optimization of the objective function in Equation (2) is computationally prohibitive because each evaluation of the likelihood function requires a d-dimensional integration, which does not have a closed form solution. Our strategy is to use Laplace's method of integration, which enables us to approximate the objective function [Eq. (2)] in closed form (up to a constant that does not involve unknown parameters):
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
& O ( \bm { \mu } , { \bf { A } } ) \approx - \frac
{ 1 } { 2 } \log \vert { \bf { A } } \vert + \frac { 1 } { { 2n
} } \mathop \sum \limits_ { i = 1 } ^n { ( { \bm { \eta } ^ { * (
i ) } } - \bm { \mu } ) ^T } { \bf { A } } ( { \eta ^ { * ( i ) }
} - \mu ) + \frac { { 1 } } { n } \mathop \sum \limits_ { i = 1 }
^n \left[ { { K^i } { { \bf { 1 } } ^T } { e^ { { \bm { \eta } ^ {
* ( i ) } } } } - { \bm { \eta } ^ { * ( i ) T } } { { \bf { x } }
^i } } \right] \\ & \quad \quad \quad + \frac { 1 } { { 2n } }
\mathop \sum \limits_ { i = 1 } ^n \log \left\vert { { \rm { diag
} } \left( { { K^i } { e^ { { \eta ^ { * ( i ) } } } } } \right) +
{ \bf { A } } } \right\vert + \lambda \mathop \sum \limits_ { j
\ne k } \; { W_ { jk } } \vert { A_ { jk } } \vert , \tag { 3 }
\end{align*}
\end{document}
To obtain parameter estimates that minimize the objective function, we alternatively update \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bm{\eta} ^{* ( i ) }}$$
\end{document} for fixed \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( \bm{\mu} , { \bf{A}} )$$
\end{document} using Newton's method, and update \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\bm{\mu}$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{A}}$$
\end{document} given \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bm{\eta} ^{* ( i ) }}$$
\end{document} using an ADMM algorithm (Boyd et al., 2011). Finally, the tuning parameter \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda$$
\end{document}, which controls the sparsity of the inferred network, is chosen by the extended Bayesian information criterion (eBIC) criterion (Chen and Chen, 2008). Details about the approximation and optimization of the objective function can be found in Supplementary Data.
2.3. Simulation studies
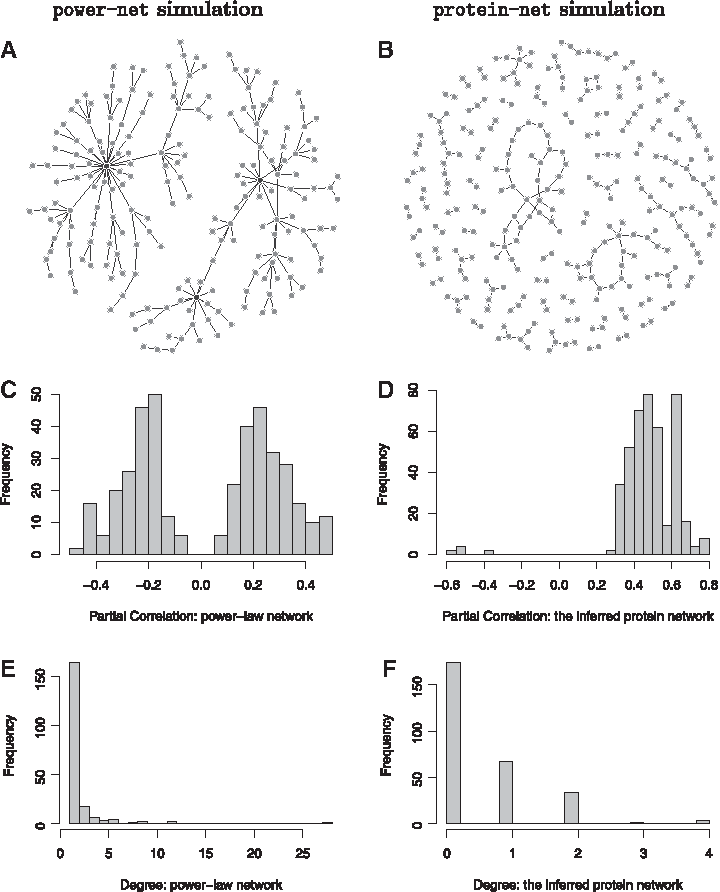
We conduct a series of simulation studies to compare the PLN and GGM approaches. Counts data are generated in the following way: we start with an assumed graph, which either follows a power-law network (Newman, 2003) or an inferred protein network (Wu et al., 2013). The mean expression level, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mu$$
\end{document}, and the concentration matrix, A, are generated according to the connectivity of the graph and a prespecified distribution that controls the signal-to-noise ratio (Fig. 1C). Next, the latent expression level \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( \bm{\eta} )$$
\end{document} and the observed counts \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \bf{x}}$$
\end{document} are simulated according to the PLN model described in Section 2.1. The simulation settings are summarized in Table 1; the details of each simulation can be found in Supplementary Data.
(A) The power-law network. (B) The protein network. (C, D) The histograms of partial correlations for power-net simulation and protein-net simulation, respectively. (E, F) The degree distributions for power-law network and protein network, respectively.
Simulation Scenarios
Label
Network
Goal of investigation
Power-net 1
Power-net
Effects of mean and variance of the underlying expression level
Power-net 2
Power-net
Overall counts distribution
Power-net 3
Power-net
Effects of sample size
Power-net 4
Power-net
Presence of a hub gene (highly connected node)
Protein-net
Protein network
Network topology and effects of the magnitudes of partial correlations
For comparison, we apply the graphical lasso (Friedman et al., 2008) to the raw count data as well as to data under several commonly used transformations: (i) normal quantile transformation, (ii) logarithmic transformation, and (iii) square-root transformation. Note that the quantile transformation assigns the same value for ties; thus the transformed data are not guaranteed to be perfectly normal if the data include many ties. Because the counts can be 0 for low-abundance genes, we add 1 to all counts before applying the logarithm transformation.
Although studies of inferring biological networks have primarily focused on edge detection, that is, whether an edge is present or not, the signs and magnitudes of the partial correlations provide clues regarding the nature of the corresponding regulation relationships: positive or negative, strong or weak. Therefore, in addition to edge detection ability, we also compare the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \ell _2}$$
\end{document} distance between the estimated and the true partial correlations:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
d (\bm{\rho} , \hat {\bm{\rho}} ) = \sqrt { \mathop \sum
\limits_{1 \le i , j \le d} \vert { \rho ^{ij}} - {{ \hat \rho
}^{ij}}{ | ^2}}.
\end{align*}
\end{document}
2.4. Application to RNA-seq data
The Geuvadis data consist of RNA-seq assays on 462 lymphoblastoid cell lines derived from five populations: European American from Utah (CEU), Finnish (FIN), British (GBR), Toscani (TSI), and African (Yoruban from Nigeria, YRI) in the 1000 Genomes Project (Lappalainen et al., 2013). We prefilter the data to focus on the 275 genes that overlap with the inferred protein network (Wu et al., 2013). To apply the PLN model and GGMs, we choose to use the RPKM (reads per kilobase of transcript per million mapped reads.), instead of the raw reads, primarily for two reasons: first, RPKM removes some systematic effects such as the batch effects and is the commonly shared form of data. Moreover, it is not always possible to reconstruct the raw read counts from RPKM. Second, empirically, we find both PLN and GGMs lead to more interpretable networks when applied to RPKM reads than to the raw reads. Three existing GGMs are used to analyze the quantile-transformed data on RPKM: (i) neighborhood selection (Meinshausen and Bühlmann, 2006), (ii) space (Peng et al., 2009), and (iii) graphical lasso (Friedman et al., 2008). The RNA-seq expression networks are inferred separately in European (CEU, FIN, GBR, and TSI; total \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\rm N} = 373$$
\end{document}) and African (YRI, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\rm N} = 89$$
\end{document}) populations. For the PLN model, the tuning parameters are selected by minimizing the eBIC with \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\gamma = 0.5$$
\end{document}; for the three GGMs, tuning parameters are adjusted so that the same number of edges are selected by all methods.
Since there is no known truth to evaluate the performance of these methods on real data, we use the modularity of the constructed network as a surrogate criterion. Each gene is assigned a functional category based on gene ontology (GO). Following Clauset et al. (2004), we define modularity as the fraction of the edges that connect two genes in the same functional category minus the expected value of such fraction if edges were distributed at random. This measure ranges between \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$- 1$$
\end{document} and 1, with higher positive values suggesting coordinated regulation of genes in related biological pathways. In practice, a modularity greater than \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.3$$
\end{document} is taken as an indicator of significant group structure in a network (Clauset et al., 2004).
3. Results
3.1. Simulation studies
We carry out simulation studies to assess the performance of the PLN model; as comparisons, we compare this model with graphical lasso applied to the raw or transformed count data (Friedman et al., 2008). Other GGM methods, such as neighborhood selection and space (Meinshausen and Bühlmann, 2006; Peng et al., 2009), are also investigated, both of which show similar results as graphical lasso (results not shown).
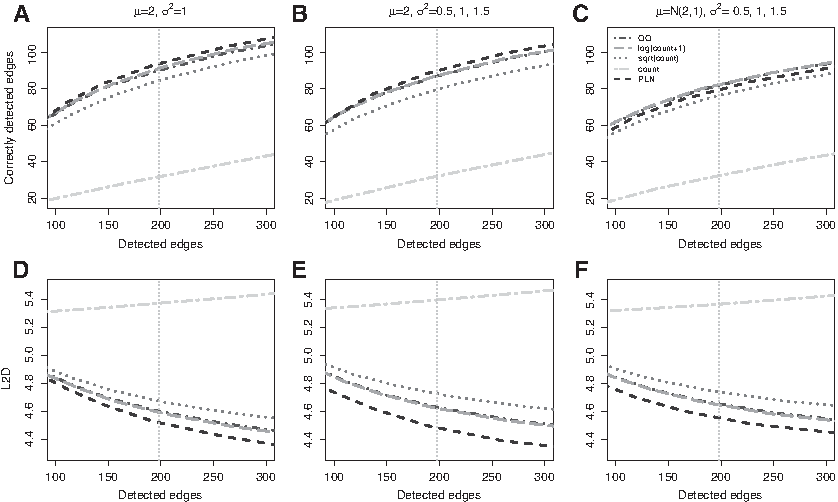
Figures 2 and 3 report the number of correctly detected edges versus the number of total detected edges, averaged across 50 independent replicates with sample size \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n = 100$$
\end{document}. A common pattern shared by all methods is that the power of edge detection improves with the increase of the overall abundance of gene expression levels (large \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _j}$$
\end{document}). When the observed counts are very low with many 0's (small \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \mu _j}$$
\end{document}), there is little information and all methods perform poorly. Measured in terms of sensitivity and specificity of edge detection, PLN performs comparably to graphical lasso on logarithm- or quantile-transformed data. All methods perform significantly better than applying graphical lasso to the raw counts directly. In addition, the performance of graphical lasso on raw or square-root transformed counts is highly sensitive to the skewness of the data and deteriorates as the variance increases (Fig. 3B, D). In contrast, PLN and graphical lasso with quantile or logarithm transformations are reasonably robust to extreme values in the data; in fact, for a fixed mean expression level, the edge-detection power of these methods improves for more skewed data with higher variance (Fig. 3B, D). Finally, Figure 2 indicates that PLN outperforms all the other methods in terms of estimating the magnitudes of the partial correlations, measured by entry-wise \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \ell _2}$$
\end{document} distance.
Power-net simulation 1. (A–C) The number of total detected edges (x-axis) and the number of correctly detected edges (y-axis) under the three scenarios of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( { \mu _j} , { \Sigma _{jj}} )$$
\end{document}. (D–F)\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \ell _2}$$
\end{document} distance (y-axis) between the estimated and the true partial correlations under the three scenarios of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( { \mu _j} , { \Sigma _{jj}} )$$
\end{document}.
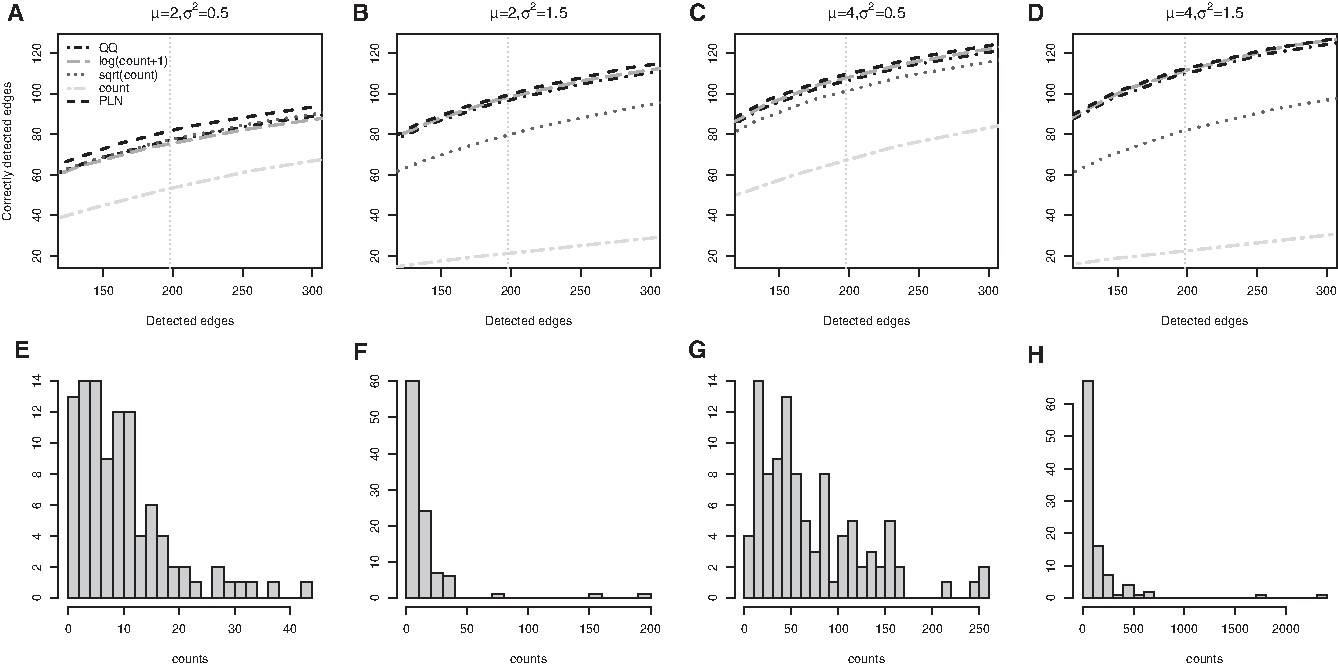
Power-net simulation 2. (A–D) The number of total detected edges (x-axis) and the number of correctly detected edges (y-axis) when \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( { \mu _j} , { \Sigma _{jj}} )$$
\end{document} are fixed at \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 2 , 0.5 )$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 2 , 1.5 )$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 4 , 0.5 )$$
\end{document}, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 4 , 1.5 )$$
\end{document} respectively. (E–H) Histograms of Poisson log-normal (PLN) counts when \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( { \mu _j} , { \Sigma _{jj}} )$$
\end{document} are fixed at \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 2 , 0.5 )$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 2 , 1.5 )$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 4 , 0.5 )$$
\end{document}, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 4 , 1.5 )$$
\end{document}, respectively.
With varying sample sizes of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n = 50$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$n = 200$$
\end{document}, not surprisingly, the performance of all methods improves with increased sample size. At a smaller sample size, PLN has more prominent advantage in detecting correct edges, whereas at a larger sample size, PLN produces comparatively more accurate estimates of partial correlations (Supplementary Fig. S1).
It has been previously observed that gene expression in a cell follows a hierarchical structure, in which the expression levels of a few genes influence the expression levels of many other genes (Morley et al., 2004). In a graph, such a “master regulator” that appears as a hub, a node is connected with many other nodes. There is an increasing interest in identifying master regulators as they play central roles in gene regulation. To investigate the ability of detecting such hub nodes, we examine the edge-detection performance in two subnetworks of the power-law network. The underlying graphs are shown in Figure 4A and B: in one subnetwork, any node is connected to at most 12 other nodes (degree \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\le 12$$
\end{document}) and there is no prominent hub; in contrast, the second subnetwork features a hub node of degree 28. Figure 4C and D compares the performance of these methods for the two subnetworks separately. Although PLN and graphical lasso with logarithm and quantile transformations perform similarly on the first subnetwork, the advantage of PLN in edge detection is more prominent in the presence of a hub node in the second subnetwork.
Power-net simulation 4: The performance on two subnetworks of the power-law network. \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( { \mu _j} , { \Sigma _{jj}} )$$
\end{document} are fixed at \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 2 , 1 )$$
\end{document}; methods are applied to the two subnetworks (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$d = 100$$
\end{document} each) separately. (A) Subnetwork 1 has no prominent hub. (B) Subnetwork 2 has 1 hub gene with 28 edges. (C, D) The number of total detected edges (x-axis) and the number of correctly detected edges (y-axis) for subnetwork 1 and subnetwork 2, respectively.
We next perform simulation according to a network that we have previously constructed in a high-throughput proteomics study, varying signal strength in terms of magnitudes of the partial correlations (Wu et al., 2013). Again, all methods, except for applying graphical lasso to raw counts, perform competitively; the advantage of PLN becomes more apparent when the signals are weak (Supplementary Fig. S2C, F).
3.2. Application to RNA-seq data
Figure 5 and Supplementary Figure S3 display the inferred networks on the Geuvadis data using four different methods: PLN, neighborhood selection (MB), space, and graphical lasso; except for PLN, all other methods are applied to quantile-transformed data. All methods are tuned such that the number of detected edges is the same: 207 for EUR and 97 for YRI. We observe that the network inferred by PLN tends to be more concentrated and hub like; in other words, fewer genes are connected by at least one edge, but these connected genes tend to have higher degrees than those in the networks constructed by other methods. In EUR, modularities are \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.32$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.36$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.37$$
\end{document}, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.43$$
\end{document} for MB, space, graphical lasso, and PLN, respectively. In YRI, the corresponding modularities are \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.37$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.50$$
\end{document}, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.57$$
\end{document}, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0.57$$
\end{document} for these four methods. We emphasize that the knowledge of functional annotation is not used in constructing the network. Therefore, the enrichment of edges connecting genes in the same GO category provides independent support that these networks recapitulate some degree of functional organization. Using this measure, the proposed PLN performs favorably compared with GGMs. Connected genes that do not fall in the same GO category may implicate uncharacterized gene function or incomplete pathways, thus offering candidates for future investigation. Moreover, we further compared hub genes between EUR and YRI networks inferred by PLN. Among top 10 high-degree genes, there were three genes (HSPA5, MANF, and PPIB) that are in common and two of them (HSPA5 and PPIB) belong to GO category; “protein transport, modification, and folding” also appear as high-degree genes in the protein network (Wu et al., 2013).
Application: The inferred networks on European population \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( n = 373 )$$
\end{document}. An edge is a solid line according to the gene ontology (GO) category if the two connecting nodes belong to the same category; otherwise, the edge is a dashed line. The number of detected edges is 207 for the PLN model and is matched to be the same for the three Gaussian graphical models (MB, space, and glasso).
4. Discussion
We have developed a graphical model approach for identifying expression covarying networks based on high-throughput RNA-seq read count data. The proposed model is hierarchical and models the observed counts as Poisson random variables with random mean parameters that follow a multivariate log-normal distribution. Conditional dependency of the underlying unobserved expression levels is then inferred from the inverse covariance matrix of the multivariate Gaussian distribution, which is estimated based on maximizing a penalized likelihood objective function. Simulation studies demonstrate that this PLN model offers several advantages over existing methods: first, PLN improves the power in edge detection compared with GGMs, especially when the sample size is small and/or the dependency is weak; second, by explicitly modeling the counts, the PLN model produces more accurate estimates of the concentration matrix, measured by the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \ell _2}$$
\end{document} distance loss; third, in simulation and analysis of real RNA-seq data, the PLN model achieves greater sensitivity in detecting edges connected to hub genes, thus facilitating the detection and investigation of master regulators.
Using a PLN distribution to model RNA-seq counts also allows for overdispersion across biological replicates, a phenomena widely observed for RNA-seq count data. In fact, the PLN model is closely related to the popular negative binomial model used in differential expression analysis for RNA-seq data (Robinson and Smyth, 2008). A negative binomial distribution can be thought of the distribution of a Poisson random variable with the mean parameter drawn from a gamma distribution. Thus, whereas the negative binomial model assumes that the underlying gene expression follows a gamma distribution, the PLN model proposed here uses a log-normal distribution instead.
Our PLN model is also related to the work of Gallopin et al. (2013), which found that a hierarchical PLN model provides a better fit of the count data, compared with Gaussian and other existing methods. The main distinction is that Gallopin et al. (2013) fit a linear model, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\eta _j^i = \sum \nolimits_{j^{\prime} \ne j} \;{ \beta _{jj^{\prime} }} \tilde x_{j^{\prime} }^i$$
\end{document}, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$x_j^i \sim$$
\end{document} Poisson(\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\exp ( \eta _j^i )$$
\end{document}) is the count of gene j for sample i and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde x$$
\end{document} is a standardization of logarithm-transformed data. In contrast, the PLN model proposed here models \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\bm{\eta}$$
\end{document} through \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\bm{\eta} \sim N ( \bm{\mu} , \bm{\Sigma} )$$
\end{document}. Another important distinction between this work and the work by Robinson and Smyth (2008) and Gallopin et al. (2013) is that our goal is to infer expression networks based on RNA-seq data, rather than identifying differentially expressed genes using RNA-seq data. Therefore, we need to model the joint distribution of the read counts rather than only inspecting their marginal distributions. The PLN model directly links to GGM through the joint distribution of the logarithm of the underlying unobserved expression levels, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\bm{\eta}$$
\end{document}, while sharing flexibility of the PLN distribution in modeling the RNA-seq counts.
On analyzing the Geuvadis RNA-seq data, PLN infers more hub-like genes and the degrees of the nodes are higher than GGMs such as MB, space, or graphical lasso. Although it is difficult to compare the performance of these methods in the real data example because of the lack of ground truth, we note that a substantial fraction of edges detected by any of the four methods connect genes with related biological functions, with PLN performing favorably measured by this modularity criterion.
This work is supported by U.S. NIH grants R01 GM073059 (Y.C, M.C., H.T.) and R01 GM082802 (J.P.), NSF grants DBI-0820854 (J.P.), and DMS-1148643 (J.P.). Y.C. was also supported by NIH T32 HG000044.
Author Disclosure Statement
No competing financial interests exist.
References
1.
AllenG.I., and LiuZ.2013. A local poisson graphical model for inferring networks from sequencing data. IEEE Trans. Nanobioscience, 12, 189–198.
2.
BoydS., ParikhN., ChuE., et al.2011. Distributed optimization and statistical learning via the alternating direction method of multipliers. Found. Trends Mach. Learn. 3, 1–122.
3.
ChenJ., and ChenZ.2008. Extended bayesian information criteria for model selection with large model spaces. Biometrika, 95, 759–771.
4.
ClausetA., NewmanM.E., and MooreC.2004. Finding community structure in very large networks. Phys. Rev. E, 70, 066111.
5.
DobraA., LenkoskiA., et al.2011. Copula Gaussian graphical models and their application to modeling functional disability data. Ann. Appl.Stat. 5, 969–993.
6.
EfronB., HastieT., JohnstoneI., et al.2004. Least angle regression. Ann. Stat. 32, 407–499.
7.
FoygelR., and DrtonM.2010. Extended bayesian information criteria for Gaussian graphical models. Adv. Neural Inf. Process. Syst. 604–612.
8.
FriedmanJ., HastieT., and TibshiraniR.2008. Sparse inverse covariance estimation with the graphical lasso. Biostatistics, 9, 432–441.
9.
GallopinM., RauA., and JaffrézicF.2013. A hierarchical poisson log-normal model for network inference from RNA sequencing data. PLoS One, 8, e77503.
10.
JalaliA., RavikumarP.D., VasukiV., et al.2011. On learning discrete graphical models using group-sparse regularization. AISTATS. 378–387. Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics.
11.
LappalainenT., SammethM., FriedländerM.R., et al.2013. Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511.
12.
LiH., and GuiJ.2006. Gradient directed regularization for sparse Gaussian concentration graphs, with applications to inference of genetic networks. Biostatistics, 7, 302–317.
13.
LiS., HsuL., PengJ., et al.2013. Bootstrap inference for network construction. Ann. Appl. Stat., 7, 391–417.
14.
LiuH., HanF., YuanM., et al.2012. High-dimensional semiparametric Gaussian copula graphical models. Ann. Stat. 40, 2293–2326.
15.
LiuH., LaffertyJ., and WassermanL.2009. The nonparanormal: Semiparametric estimation of high dimensional undirected graphs. J. Mach. Learn. Res., 10, 2295–2328.
16.
MeinshausenN., and BühlmannP.2006. High-dimensional graphs and variable selection with the lasso. Ann. Stat. 34, 1436–1462.
17.
MeinshausenN., and BühlmannP.2010. Stability selection. J. R. Stat. Soc. Series B Stat. Methodol. 72, 417–473.
18.
MorleyM., MolonyC.M., WeberT.M., et al.2004. Genetic analysis of genome-wide variation in human gene expression. Nature. 430, 743–747.
19.
NewmanM.E.2003. The structure and function of complex networks. SIAM Rev. 45, 167–256.
20.
OhJ.H., and DeasyJ.O.2014. Inference of radio-responsive gene regulatory networks using the graphical lasso algorithm. BMC Bioinformatics, 15Suppl 7, S5.
21.
PengJ., WangP., ZhouN., et al.2009. Partial correlation estimation by joint sparse regression models. J. Am. Stat. Assoc. 104, 735–746.
22.
RavikumarP., WainwrightM.J., LaffertyJ.D., et al.2010. High-dimensional ising model selection using 1-regularized logistic regression. Ann. Stat. 38, 1287–1319.
23.
RobinsonM.D., McCarthyD.J., and SmythG.K.2010. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140.
24.
RobinsonM.D., and SmythG.K.2008. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics, 9, 321–332.
25.
TibshiraniR.1996. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Series B Methodol. 58, 267–288.
26.
WangP., ChaoD.L., and HsuL.2011. Learning oncogenic pathways from binary genomic instability data. Biometrics, 67, 164–173.
27.
WolfeP.1969. Convergence conditions for ascent methods. SIAM Rev. 11, 226–235.
28.
WolfeP.1971. Convergence conditions for ascent methods. II: Some corrections. SIAM Rev. 13, 185–188.
29.
WuL., CandilleS.I., ChoiY., et al.2013. Variation and genetic control of protein abundance in humans. Nature, 499, 79–82.
30.
YangE., RavikumarP.K., AllenG.I., et al.2013. On poisson graphical models. Adv. Neural Inf. Process. Syst. 1718–1726.
31.
YuanM., and LinY.2007. Model selection and estimation in the Gaussian graphical model. Biometrika, 94, 19–35.
32.
ZouH., HastieT., TibshiraniR., et al.2007. On the “degrees of freedom” of the lasso. Ann. Stat. 35, 2173–2192.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.