
Abstract
Abstract
Genome annotation is a primary step in genomic research. To establish a light and portable prokaryotic genome annotation pipeline for use in individual laboratories, we developed a Shiny app package designated as “P-CAPS” (Prokaryotic Contig Annotation Pipeline Server). The package is composed of R and Python scripts that integrate publicly available annotation programs into a server application. P-CAPS is not only a browser-based interactive application but also a distributable Shiny app package that can be installed on any personal computer. The final annotation is provided in various standard formats and is summarized in an R markdown document. Annotation can be visualized and examined with a public genome browser. A benchmark test showed that the annotation quality and completeness of P-CAPS were reliable and compatible with those of currently available public pipelines.
1. Introduction
A
To complement these issues and provide a more generally accessible genome annotation pipeline, we developed a web-based application (webApp) termed P-CAPS (Prokaryotic Contig Annotation Pipeline Server). The pipeline is managed under the ShinyR package, a webApp framework for R. P-CAPS can be easily deployed on the local Linux system equipped with various open-source bioinformatics tools for genome annotation. Users can also launch P-CAPS as their own webApp service in a Shiny server. The pipeline is maintained with an R script language without complete knowledge about HTML or JavaScript. P-CAPS lets users embed additional R scripts for their own analyses. Because the pipeline is handled directly in the web browser, users can plug external programs such as JBrowse (Skinner et al., 2009) into P-CAPS. A graphical overview written in R markdown is provided as a summary report.
2. Description
2.1. Input
The input file of the pipeline consists of nucleotide sequences (contigs) in FASTA format with the extension “.fna,” “.fasta,” or “.fa.”
2.2. Annotation pipeline
P-CAPS is divided into two steps: (1) gene prediction and (2) functional annotation. In the gene prediction step, we implemented a consensus algorithm using two gene predictors, Prodigal (Hyatt et al., 2010) and GeneMarkS (Besemer et al., 2001), for improving the prediction accuracy. The outputs from the programs are com-pared and merged into a refined set by the consensus algorithm. The structural RNA regions are predicted by RNAmmer (Lagesen et al., 2007) for ribosomal RNAs (rRNAs) and by transfer RNA (tRNA)scan-SE (Lowe and Eddy, 1997) for tRNAs. Predicted coding sequences (CDSs) are annotated with orthologous genes based on a BLAST (Boratyn et al., 2013) search across the UniRef90 protein database (The UniProt Consortium, 2010) or other protein databases. The KEGG ORTHOLOGY (KO) number is assigned according to the transitive relations in UniProtKB (The UniProt Consortium, 2010). For the analysis of predicted CDSs at the domain level, a protein domain annotation procedure is implemented in P-CAPS using InterProScan (Jones et al., 2014).
2.3. Output
P-CAPS provides the annotation result in various standard file formats such as FASTA, GFF3, and GBFF (GBK), along with a graphical summary report written in R markdown. The summary report presents basic statistics of genome annotation such as sequence length, GC ratio, and annotated feature counts. The annotation completeness by Benchmarking Universal Single-Copy Ortholog (BUSCO) (Simão et al., 2015) and the functional categorization by KO are included in the summary report (Fig. 1).
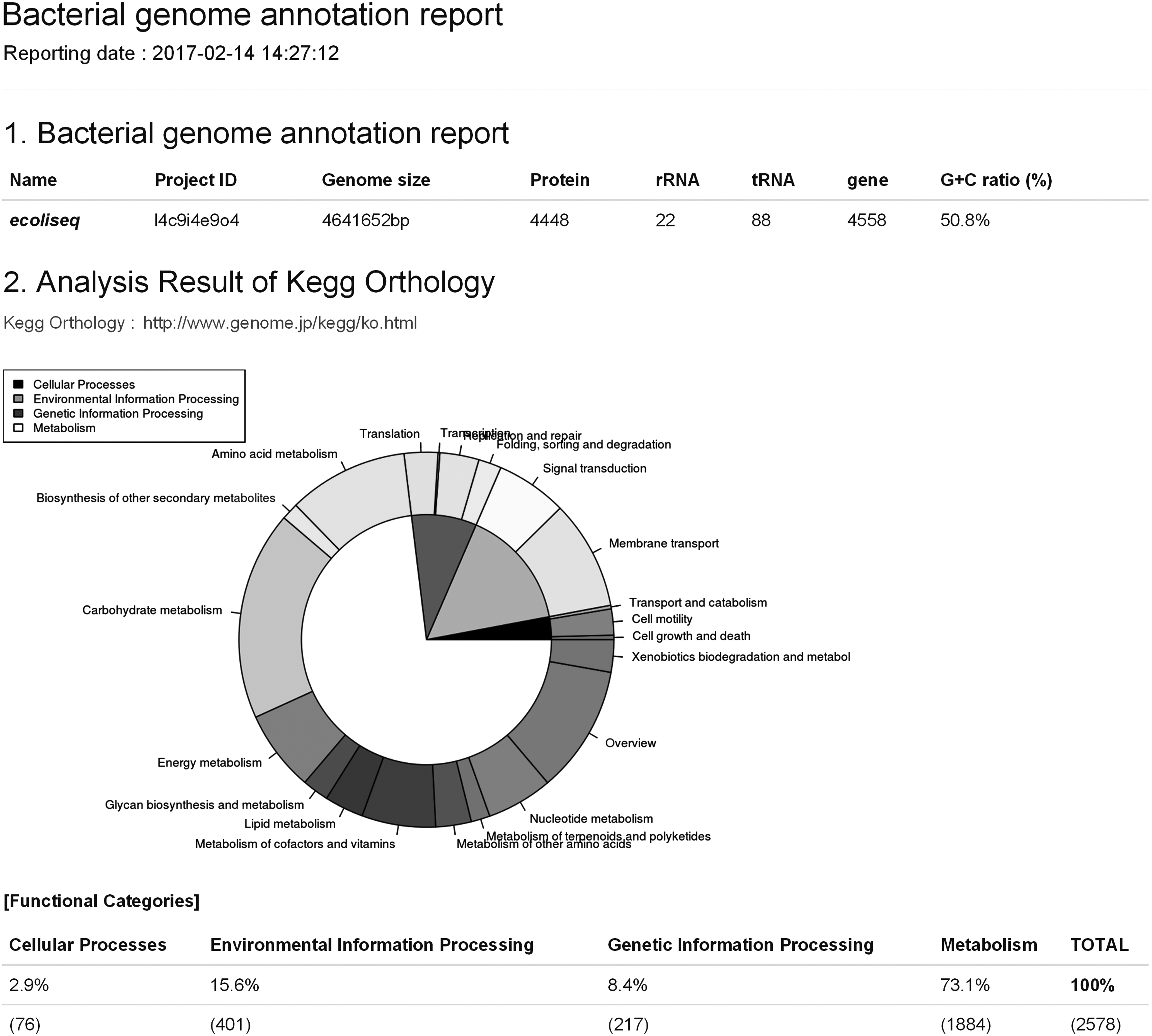
Example of a graphical summary report from P-CAPS. The summary report is created with R markdown and is downloadable in DOCX, PDF, and HTML formats. P-CAPS, Prokaryotic Contig Annotation Pipeline Server; rRNA, ribosomal RNA; tRNA, transfer RNA.
2.4. Visualization
For manual inspection of the genome annotation, the user can use JBrowse (Skinner et al., 2009), which is integrated in P-CAPS. By default, the annotation results are automatically converted into the format required to launch JBrowse. Any genome browser can then be used to visually scan the annotation results.
2.5. Availability
P-CAPS is an open source application based on the Shiny R package. P-CAPS is demonstrated and serviced at http://panflam.korea.ac.kr/pcaps/mainpage, and a stand-alone version is downloadable from https://github.com/choilab/P-CAPS. The installation guide and manual for the stand-alone version are available from https://github.com/CompSynBioLab-KoreaUniv/P-CAPS/wiki.
3. Results
To evaluate the performance of P-CAPS, we benchmarked the package against publicly available annotation programs (RAST and Prokka) using NCBI (National Center for Biotechnology Information) reference genomes from various taxa: Escherichia coli str. K-12 substr. MG1655 (GCA_000005845.2), Lactobacillus plantarum WCFS1 (GCA_000203855.3), Bacillus subtilis subsp. subtilis str. 168 (GCA_000009045.1), Eubacterium limosum KIST612 (GCA_000152245.2), and Vibrio sp. EJY3 (GCA_000241385.1). P-CAPS showed comparable performance to the other programs for predicting major genomic features such as the CDSs, tRNAs, and rRNAs (Table 1). We checked the annotation quality according to the completeness of genome annotation using BUSCO (Simão et al., 2015). The annotations from P-CAPS and Prokka showed near completeness of the BUSCO checkup over the tested NCBI RefSeq genomes (Table 2). RAST missed a few CDSs from incongruent start sites of the RefSeq, which was due to short and fragmented protein sequence prediction (Fig. 2). The functionality of P-CAPS as a web application with a Shiny server is tested in the server of Korea University (http://panflam.korea.ac.kr/pcaps/mainpage).

Example of the missed and fragmented BUSCOs annotated by RAST. The results were not in accordance with the annotation from P-CAPS and other pipelines. The RAST annotation showed
Five complete genome sequences from NCBI GenBank were used for the benchmark test.
CDS, coding sequence; NCBI, National Center for Biotechnology Information; P-CAPS, Prokaryotic Contig Annotation Pipeline Server; RAST, Rapid Annotations using Subsystems Technology; rRNA, ribosomal RNA; tRNA, transfer RNA.
The following BUSCO databases were used according to the taxon of each genome: Enterobacteriales odb9 for E. coli str. K-12 substr. MG1655, Lactobacillales odb9 for L. plantarum WCFS1, Bacillales odb9 for B. subtilis subsp. subtilis str. 168, Clostridia odb9 for E. limosum KIST612, and Gamma proteobacteria odb9 for Vibrio sp. EJY3.
BUSCO, Benchmarking Universal Single-Copy Ortholog.
Footnotes
Acknowledgments
This work was supported by Institute of Life Science and Natural Resources and the BK21plus program at Korea University.
Author Disclosure Statement
No competing financial interests exist.