The alignment of three or more protein or nucleotide sequences is known asMultiple Sequence Alignment problem. The complexity of this problem increases exponentially with the number of sequences; therefore, many of the current approaches published in the literature suffer a computational overhead when thousands of sequences are required to be aligned. We introduce a new approach for dealing with ultra-large sets of sequences. A two-level clustering method is considered. The first level clusters the input sequences by using their biological composition, that is, the number of positive, negative, polar, special, and hydrophobic amino acids. In the second level, each cluster is divided into different clusters according to their similarity. Then, each cluster is aligned by using any method/aligner. After aligning the centroid sequences of each second-level cluster, we extrapolate the new gaps to each cluster of sequences to obtain the final alignment. We present a study on biological data with up to ∼100,000 sequences, showing that the proposed approach is able to obtain accurate alignments in a reduced amount of time; for example, in>10,000 sequences datasets, it is able to reduce up to ∼45 times the required runtime of the well-known Kalign.
1. Introduction
Multiple Sequence Alignment (MSA) (Bacon and Anderson, 1986) is the simultaneous alignment of three or more protein/nucleotide sequences. In Doolittle (1981), the authors considered the MSA problem as a crucial step for inferring phylogenetic relationships among species. In addition, an accurate alignment helps to study proteins and nucleotides with a strong biological significance level (Feng and Doolittle, 1987).
Given a set of N unaligned sequences \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$S: \{ {s_1} , {s_2} , \ldots , {s_N} \} $$
\end{document} defined over an alphabet \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Sigma$$
\end{document} (amino-acid or nucleotide alphabet), the MSA of this set is defined as \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$S {^\prime} : \{ {s {^\prime} _1} , {s {^\prime} _2} , \ldots , {s {^\prime} _N} \} $$
\end{document}, where all the sequences are of equal length (L) and defined over the alphabet \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Sigma \cup \{ - \} $$
\end{document}, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$-$$
\end{document} refers to the gap symbol. In addition, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${s {^\prime} _i} = {s_i}$$
\end{document} for all i, when any gaps are removed from \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${s {^\prime} _i}$$
\end{document}. In the following, we present an example of MSA:
As can be observed, the alignment is a matrix, where the rows are the sequences and the columns are the aligned symbols. A column with all gaps is not allowed, so each column contains at least one symbol of the original alphabet (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Sigma$$
\end{document}). To obtain the optimal alignment among the N sequences, we require a computational complexity of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {L^N} )$$
\end{document}, that is, the complexity suffers an exponential growth as the number of sequences increases.
In the literature, we find progressive methods that start building up a guide tree and using it to align the input set of sequences (Hogeweg and Hesper, 1984). In general, the progressive methods have a computational complexity of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {N^2} )$$
\end{document}; therefore, their performance is acceptable when dealing with alignments of a few thousand sequences of moderate length, but their performance is considerably reduced with bigger datasets. The consistency-based methods were introduced by Notredame et al. (2000) and their complexity is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {N^3}{L^2} )$$
\end{document}, which allows us to align a few hundreds of sequences. The last group of methods published in the literature uses a refinement stage, and we refer to them as iterative refinement methods. The complexity of these methods is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {N^2}L + N{L^2} )$$
\end{document} without the refinement stage, and it adds an \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {N^3}L )$$
\end{document} term if it is considered. Finally, other authors have dealt with the MSA problem by using stochastic approaches, such as genetic algorithms (Notredame and Higgins, 1996; Gondro and Kinghorn, 2007) and multiobjective evolutionary algorithms (Ortuño et al., 2013; Rubio-Largo et al., 2016b,c). The main disadvantage of these methods is their prohibitive running time.
In recent literature, some efforts have been conducted to solve sets of thousands of sequences, such as Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} (Sievers et al., 2011) and Practical Alignment by using Sate and TrAnsitivity (PASTA) (Mirarab et al., 2015). On the one hand, Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} uses seeded guide trees and profile–profile techniques to generate alignments. On the other hand, PASTA makes use of transitivity to align sequences on a guide tree and also improves the alignment of very distantly related sequences and remote homology detection.
In this article, we introduce a new approach for dealing with ultra-large sets of sequences (thousands of sequences). A two-level clustering method is proposed. The first level clusters the input sequences by using their biological composition, that is, the number of positive, negative, polar, special, and hydrophobic amino acids. In the second level, each group obtained in the first level is divided into different clusters according to their similarity. Then, each second-level cluster is aligned by using any method/aligner. From each aligned cluster, we pick its centroid sequence (seed sequence) and perform an alignment. Finally, we extrapolate the new inserted gaps to each cluster of sequences to obtain the final alignment. It is freely available for downloading at (Rubio-Largo et al., 2017).
We present a study on biological data with up to ∼100,000 sequences (HomFam benchmark), showing that the proposed approach is able to obtain accurate alignments in a reduced amount of time. In the study, we test the performance of our approach by using the following aligners: Muscle (Edgar, 2004b), Kalign (Lassmann et al., 2009), and multiple alignment using Fast Fourier transformation (MAFFT) (Katoh et al., 2002). In addition, they are compared with two well-known aligners that are designed for ultra-large sets of sequences: Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} and PASTA.
The rest of the work is organized as follows. In Section 2, we detail the steps of the approach. A study on biological data is presented in Section 3 to demonstrate the effectiveness of the proposed approach. Finally, conclusions and future lines of work appear in Section 4.
2. Methods
This section describes the proposed approach. An illustrative representation of the method is presented in Figure 1, which will be helpful for a better understanding of the steps conducted by the method when aligning an input set of unaligned sequences. Given an input set of N unaligned sequences, the procedure of the approach is as follows:
Step 1. Analyze the biological composition of each unaligned sequence to obtain a matrix of characteristics (A) (Rubio-Largo et al., 2016a). For each unaligned sequence in the input set, count the number of positive (R, H, and K), negative (D and E), polar (S, T, N, and Q), special (C, U, G, and P), and hydrophobic (A, V, I, L, M, F, Y, and W) amino acids that it contains. Therefore, A is an \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N \times 5$$
\end{document} matrix.
Step 2. By using k-means clustering, classify the N unaligned sequences into k1 clusters according to the matrix of biological characteristics (A) computed in the previous step. The output of the k-means is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ {c_1} , {c_2} , \ldots , {c_{k1}} \} $$
\end{document}, where the notation \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\vert {c_i} \vert$$
\end{document} indicates the number of sequences contained in cluster i. The computational complexity of k-means is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {N^{5*{k_1}}} )$$
\end{document}, but we have used a linear approximation, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( N )$$
\end{document}.
Step 3. For each cluster in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ {c_1} , {c_2} , \ldots , {c_{k1}} \} $$
\end{document}, use the UCLUST algorithm (Edgar, 2010) to divide it into clusters. UCLUST is a greedy heuristic-based clustering algorithm that employs USEARCH (Edgar, 2004a) as a subroutine to assign sequences to clusters. According to Edgar (2010), in UCLUST, an initially empty database is created, which is extended as input sequences are processed. The database contains exactly one representative sequence for each cluster, known as its seed. Each input sequence (s) is compared with the current database by using USEARCH. If a matching seed is found, s is assigned to the corresponding cluster and the database remains unchanged; otherwise, s is added to the database, becoming the seed of a new cluster. A match is defined as a global alignment of the query to a seed with an identity exceeding a pre-set threshold (ID). The computational complexity of UCLUST is quasilinear: \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {N^{1.2}} )$$
\end{document} (Edgar, 2010). Therefore, the total complexity of this step is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\sum \nolimits_{i = 1}^{{k_1}} O ( \vert {c_i}{ \vert ^{1.2}} )$$
\end{document}.
Step 4. After applying the UCLUST algorithm to the k1 clusters, we obtained a new set of clusters: \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ {c_1} , {c_2} , \ldots , {c_{k2}} \} $$
\end{document}. For each cluster in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ {c_1} , {c_2} , \ldots , {c_{k2}} \} $$
\end{document}, use any aligner method to align the set of sequences contained in the cluster. The complexity of Step 4 depends on the complexity of the aligner used. For example, if the complexity of a progressive-based aligner is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( {N^2} )$$
\end{document}, the complexity of the current step is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\sum \nolimits_{i = 1}^{{k_2}} O ( \vert {c_i}{ \vert ^2} )$$
\end{document}.
Step 5. From each isolated alignment, extract the aligned seed sequence, replace the gap symbol by the undefined symbol X, and create a set of k2 sequences. Align the set of k2 seed sequences and obtain the seed alignment. Following the previous example, if we consider a progressive-based aligner, the computational complexity is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$O ( ( {k_2}{ ) ^2} )$$
\end{document}.
Step 6. From the seed alignment, extract one by one the aligned sequences and extrapolate the new inserted gaps to their related cluster alignment. In this way, the total length of the k2 isolated alignment will be the same. Finally, merge the extrapolated k2 alignments and create the final alignment. The complexity of the final step is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\sum \nolimits_{i = 1}^{{k_2}} O ( \vert {c_i} \vert )$$
\end{document}.
Illustrative view of the new multiple sequence alignment approach.
In the proposed approach, any aligner published in the literature can be used. The k-means clustering implementation was taken from Curtin et al. (2013). We have used \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$ { k_1 } = \left\lceil { \frac { N } { { 500 } } } \right\rceil + 1$$
\end{document}; therefore, the k-means approach always divides the N input sequences into a minimum of two clusters. Following the advice of Edgar (2010), the pre-set threshold (ID) used in UCLUST was set to 0.5.
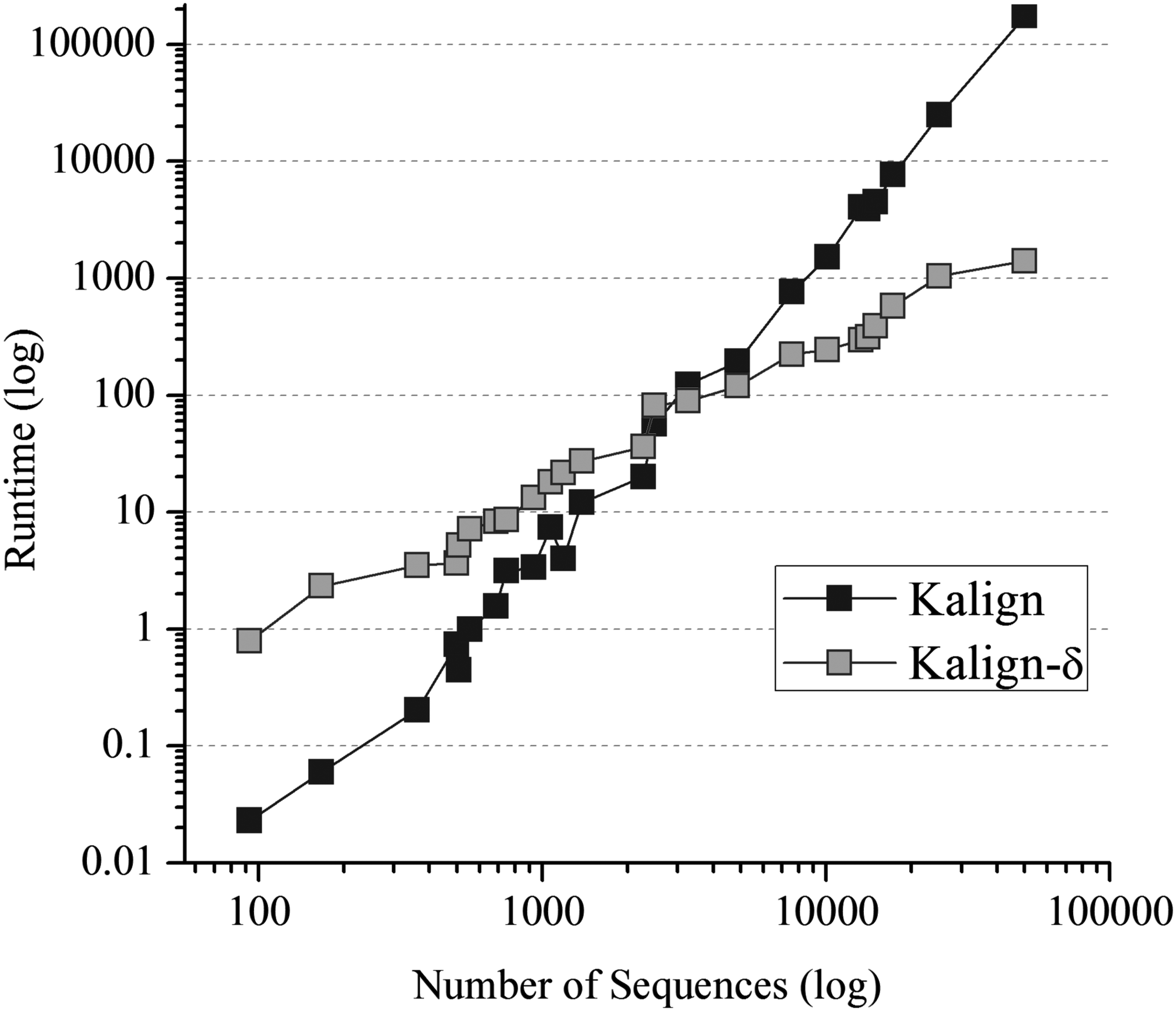
As can be observed, Steps 4 and 5 contribute more to the total computational complexity of our approach; however, the total computational complexity has been considerably reduced compared with the traditional aligner. In Figure 2, we present a runtime comparison between the progressive-based aligner Kalign (Lassmann et al., 2009) and our approach with the Kalign version; we refer to this as Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}. As can be observed, the differences of runtime rise quadratically as the number of sequences increases, showing significant differences with a number of sequences greater than 3000.
Runtime comparison (in seconds) between Kalign and Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}.
3. Results
Since the proposed methodology was designed to reduce the time complexity of any aligner when dealing with ultra-large sets of sequences, we have used an ultra-large real biological benchmark: HomFam (Sievers et al., 2011). The HomFam benchmark contains 89 HOMSTRAD families as reference MSAs and their corresponding Pfam sequences to be aligned. Following Sievers et al. (2011), the HomFam benchmark was divided into three groups: small (0\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$<$$
\end{document}N\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\le$$
\end{document}3000), medium (3000\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$<$$
\end{document}N\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\le$$
\end{document}10,000), and large (N\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\ge$$
\end{document}10,000), where N is the number of sequences.
The methods involved in the comparison are as follows:
• Kalign v2.04 (Lassmann et al., 2009). It was run with default parameter configuration.
• Muscle v3.8.31 (Edgar, 2004b). This aligner uses its default parameter configuration when \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N <$$
\end{document} 2000 sequences. For larger sets, Muscle is run with the flag -maxiters 2 to finish the alignments in reasonable times.
• MAFFT v7.305b (Katoh et al., 2002). It was run with the –auto flag. This flag chooses to run a slow, consistency-based program (L-INS-i) when the number and lengths of sequences is small. When the numbers exceed inbuilt thresholds, a conventional progressive aligner is used (FFT-NS-2). For very large datasets, a very fast guide tree calculation is then used.
• Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} v1.2.0 (Sievers et al., 2011). The –auto flag was used to allow Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} to automatically set the execution options depending on the input set of sequences. In addition, it was run by using a single thread (–threads = 1).
• PASTA v1.6.4 (Mirarab et al., 2015). This aligner divides the input set of sequences into smaller subsets, each of which is aligned by using an external alignment tool (default is MAFFT). These subset alignments are then pairwise merged (by default using Opal) and, finally, the pairwise merged alignments are merged into a final alignment by using transitivity. PASTA is run by using its default parameter configuration and single thread (–num-cpus = 1).
• The proposed method (-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}). The approach proposed in this article was tested by using three different aligners: Kalign v2.04, Muscle v3.8.31, and MAFFT v7.305b; we refer to them as Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, and MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, respectively.
All the aligners were run on an AMD Opteron(tm) Processor 6176 @ 2.30 GHz with 64 GB RAM.
To quantify the agreement between the true alignment and the alignments obtained by the aligners, two measures of accuracy are commonly employed in the literature. These assessment metrics are: Q and total column (TC) (Edgar, 2004b). On the one hand, Q (quality) indicates the number of correctly aligned residue pairs divided by the number of residue pairs in the reference alignment. On the other hand, the TC score is the number of correctly aligned columns divided by the number of columns in the reference alignment. The alignment accuracies were computed by using the qscore program (Edgar, 2015).
In Table 1, we present the Q and TC scores (in %) obtained by the different aligners in the three groups [small, medium, and large (Yamada et al., 2016)] of the HomFam benchmark, as well as their required runtime for solving the benchmark (in minutes). In addition, Figure 3 illustrates the differences of Q and TC among the different approaches.
Illustrative comparison among the different approaches in terms of Q (%) and TC (%). TC, total column.
Comparison Among Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document}, Kalign, Muscle, MAFFT, PASTA, Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, and MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} in Terms of Q (%), TC (%), and Runtime (t, in Minutes)
As may be observed, with a small number of sequences (N < 3000), the most accurate approaches are Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} and MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}; however, although the Q and TC scores obtained by MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} are lower than those obtained by Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document}, it requires only 22 minutes to complete the 38 alignments included in the small group, whereas Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} requires 52 minutes. In terms of required runtime, MAFFT and Kalign are the two fastest approaches, requiring only around 8.5–9 minutes to finish. If we focus on the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} approaches, it can be observed that our approach does not provide any advantage for either Kalign or MAFFT in the small group. However, for Muscle, the benefits are noticeable in both alignment quality and runtime. On the one hand, in terms of Q and TC scores, Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} obtained 85.94% (Q) and 73.79% (TC) whereas Muscle achieved 81.83% (Q) and 69.02% (TC). On the other hand, it can be observed that Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} is around 13 times faster than Muscle. Comparing PASTA and MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} (both make use of MAFFT), it can be observed that MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} overcomes PASTA in alignment quality and is around 40 times faster.
Analyzing the results obtained by the aligners with a medium number of sequences \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( 3000 < N \le 10 , 000 )$$
\end{document}, it can be observed that the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} approaches of Kalign, Muscle, and MAFFT obtain better results than their original versions. Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} obtains similar Q and TC scores to Kalign; however, it only takes 39 minutes to align the 32 files, whereas Kalign requires 184 minutes. Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} clearly overcomes Muscle, and it is able to improve Q and TC by +10.93% and +17.4%, respectively. Finally, MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} obtains the highest values of Q and TC, overcoming the results of the other approaches involved. Compared with Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} and PASTA, MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} obtains 91.25%/78.72% (Q/TC) in 75 minutes, whereas PASTA and Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} obtain 82.92%/73.02% in 3625 minutes and 86.94%/72.08% in 351 minutes.
With a large number of sequences (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N > 10 , 000$$
\end{document}), the advantages of applying the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} methodology to Kalign, Muscle, and MAFFT are also remarkable. For Kalign and Muscle, a great improvement in terms of Q and TC scores can be observed: +10.48%/+15.54% and +30.97%/+26.56%, respectively. In addition, Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} and Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} are up to 45.5 and 14.5 times faster than Kalign and Muscle. MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} and MAFFT obtain very similar values of Q and TC; however, MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} is more than 2.5 times faster than MAFFT. Analyzing the results of MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} and PASTA, we realize that MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} obtains greater Q scores than PASTA, and PASTA overcomes MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} in terms of TC. If we focus on runtime, we observe that MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} requires 197 minutes to align the 19 files of the large group, whereas PASTA needs 7944 minutes, that is, MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} is around 40 times faster than PASTA with a dataset of up to 100,000 sequences. Finally, all the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} approaches outperform Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document}.
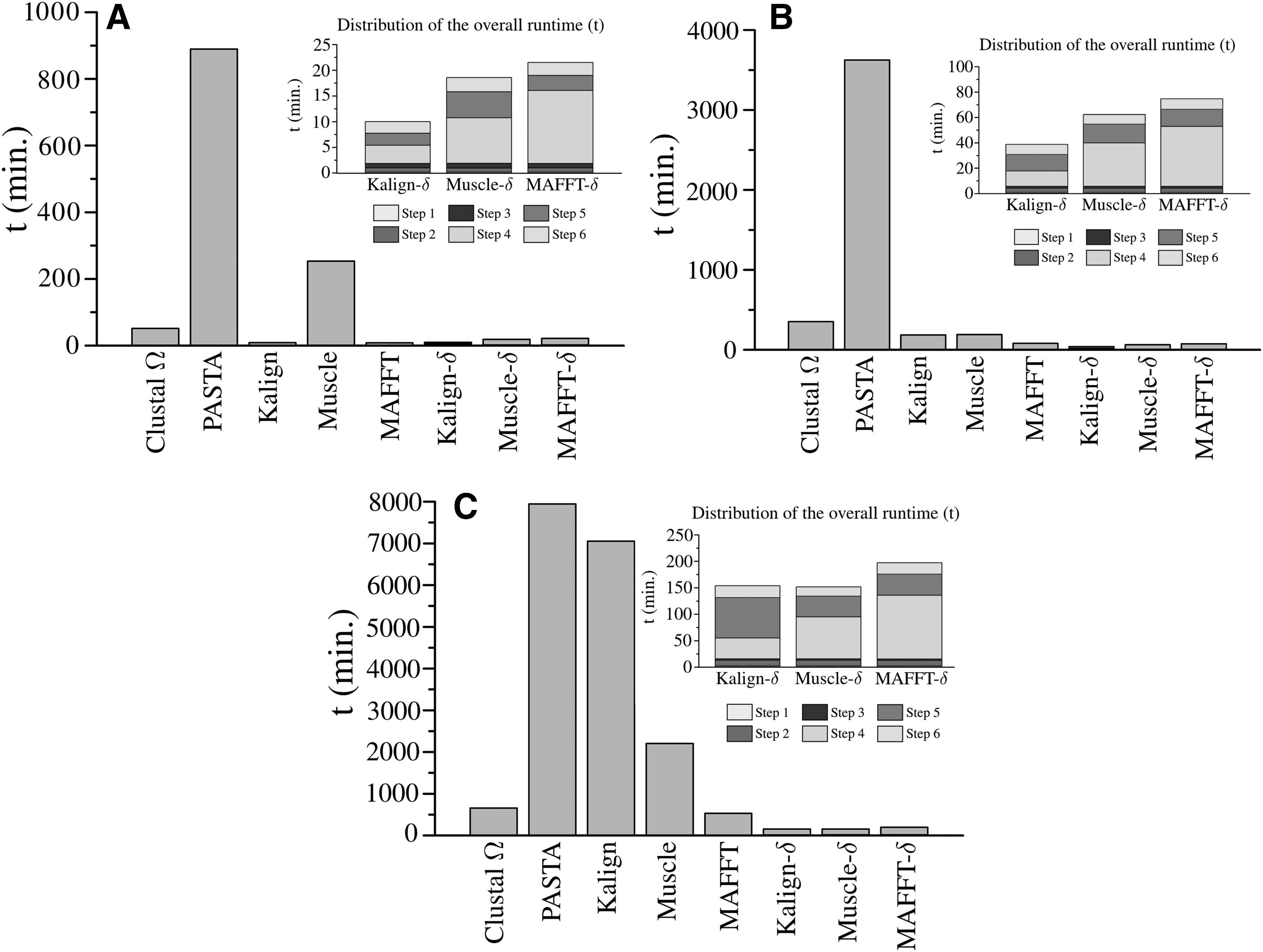
In Figure 4, we illustrate the required runtime of each aligner under study for the three groups of HomFam. In addition, we plot the runtime distribution of the three \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} approaches to show the influence of each step (Section 2).
Runtime (t) comparison among the different approaches in (A) small (0, 3000], (B) medium (3000, 10,000], and (C) large (10,000] HomFam datasets. In addition, we plot the distribution of the overall runtime spent in the different steps (Section 2) of Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, and MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}.
To ensure a certain level of statistical reliability, we have performed a pair-wise Wilcoxon signed-rank test between the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} approaches (Kalign-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, Muscle-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}, and MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document}) and the other approaches (Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document}, Kalign, Muscle, MAFFT, and PASTA). In Table 2, we present the number of datasets in which the Q and TC differences between each pair of aligners are statistically significant with a confidence level of 1% (p-value \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$<$$
\end{document}0.01). As can be observed, in the small, medium, and large groups, the differences are statistically significant in the vast majority of the datasets.
Note that at each cell, we find the following notation: a/b, where a indicates the number of datasets in which the differences between the two methods are statistically nonsignificant in terms of Q score, and b indicates the number of datasets in which the differences between the two methods are statistically nonsignificant in terms of TC score.
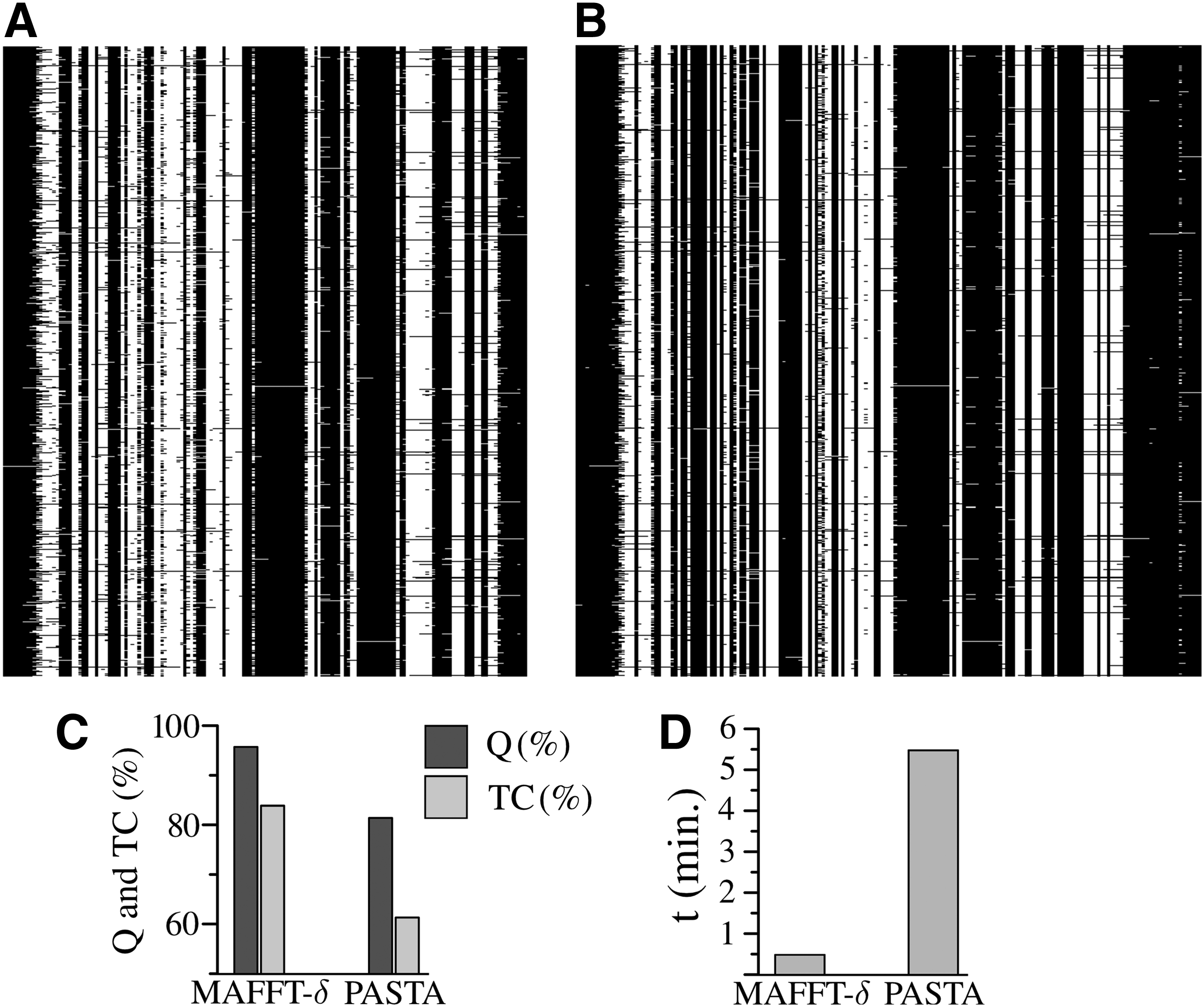
Finally, in Figure 5, as an example, we compare the alignments obtained by PASTA and MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} when dealing with the small dataset il8 (1073 sequences). As can be observed, MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} obtains better Q and TC scores than PASTA and it takes ∼30 seconds, whereas PASTA requires more than 5 minutes. Also, in Figure 5A and B, it can be observed that PASTA requires a higher number of gaps than MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} (represented in black).
Analysis of the alignment produced by (A) MAFFT-\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\delta$$
\end{document} and (B) PASTA for the small dataset il8 (1073 sequences). In (C), we compare the alignment quality in terms of Q (%) and TC (%). In (D), we present the required runtime by each method (in minutes).
4. Conclusions
This article proposes a new approach for aligning ultra-large sets of sequences (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$>$$
\end{document}10,000 sequences).
The approach uses a two-level clustering method. In the first level, it clusters the input sequences by considering their biological composition, that is, the number of positive, negative, polar, special, and hydrophobic amino acids. In the second level, each cluster obtained in the first level is divided into new clusters according to their similarity. Then, each second-level cluster is aligned by using any method/aligner. From each aligned cluster, its centroid sequence is picked (seed sequence) and an alignment is performed. After that, the new inserted gaps are extrapolated to each cluster of sequences to output a consistent alignment.
To test the performance of the proposed approach, we have used a real biological benchmark: HomFam. This benchmark contains 89 files that are divided into three groups according to the number of sequences: small (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$0 < N \le 3000$$
\end{document}), medium (\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$3000 < N \le 10 , 000$$
\end{document}), and large \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$( N \ge 10 , 000 )$$
\end{document}), where N is the number of sequences. The aligners tested within the proposed method were Kalign, Muscle, and MAFFT. They were compared against Clustal \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Omega$$
\end{document} and PASTA, two well-known aligners designed for ultra-large sets of sequences. Results show that our approach provides great benefits to these aligners with respect to both alignment quality and runtime. From the comparative study, we can conclude that the three tested aligners obtain benefits from the proposed method, highlighting a great improvement of alignment quality and runtime for Kalign and Muscle when \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N > 3000$$
\end{document} sequences.
As future work, we intend to perform a comprehensive study among very different aligners and other real biological benchmarks. In addition, the parallelization of the proposed heuristic is an immediate line of future work, because it will allow us to align ultra-large sets of sequences in seconds.
Footnotes
Acknowledgments
This work was partially funded by the AEI (State Research Agency, Spain) and the ERDF (European Regional Development Fund, EU), under the contract TIN2016-76259-P (PROTEIN project). Á.R.-L. is supported by the post-doctoral fellowship SFRH/BPD/100872/2014 granted by Fundação para a Ciência e a Tecnologia (FCT), Portugal.
Author Disclosure Statement
No competing financial interests exist.
References
1.
Rubio-LargoA., VanneschiL., CastelliM., et al. (2017). Delta Software Available at: arco.unex.es/arl/delta.zip (date: 2017).
2.
BaconD.J., and AndersonW.F. (1986). Multiple sequence alignment. J. Mol. Biol., 191, 153–161.
3.
CurtinR.R., ClineJ.R., SlagleN.P., et al. (2013). mlpack: A scalable C++ machine learning library. J. Mach. Learn. Res., 14, 801–805.
4.
DoolittleR. (1981). Similar amino acid sequences: Chance or common ancestry?. Science, 214, 149–159.
5.
EdgarR.C. (2004a). Local homology recognition and distance measures in linear time using compressed amino acid alphabets. Nucl. Acids Res., 32, 380–385.
6.
EdgarR.C. (2004b). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res., 32, 1792–1797.
7.
EdgarR.C. (2010). Search and clustering orders of magnitude faster than blast. Bioinformatics, 26, 2460–2461.
8.
EdgarR.C. (2015). QSCORE: A quality scoring program that compares two multiple sequence alignments. Available at: www.drive5.com/qscore
9.
FengD., and DoolittleR. (1987). Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol., 25, 351–360.
10.
GondroC., and KinghornB.P. (2007). A simple genetic algorithm for multiple sequence alignment. Genet. Mol. Res., 6, 964–982.
11.
HogewegP., and HesperB. (1984). The alignment of sets of sequences and the construction of phyletic trees: An integrated method. J. Mol. Biol., 20, 175–186.
12.
KatohK., MisawaK., KumaK., et al. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucl. Acids Res., 30, 3059–3066.
13.
LassmannT., FringsO., and SonnhammerE.L.L. (2009). Kalign2: High-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucl. Acids Res., 37, 858–865.
14.
MirarabS., NguyenN., GuoS., et al. (2015). PASTA: Ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. J. Comput. Biol., 22, 377–386.
15.
NotredameC., and HigginsD.G. (1996). SAGA: Sequence alignment by genetic algorithm. Nucl. Acids Res., 24, 1515–1524.
16.
NotredameC., HigginsD.G., and HeringaJ. (2000). T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol., 302, 205–217.
17.
OrtuñoF.M., ValenzuelaO., RojasF., et al. (2013). Optimizing multiple sequence alignments using a genetic algorithm based on three objectives: Structural information, non-gaps percentage and totally conserved columns. Bioinformatics, 29, 2112–2121.
18.
Rubio-LargoA., VanneschiL., CastelliM., et al. (2016a). A characteristic-based framework for multiple sequence aligners. IEEE Trans. Cybern.1–11. DOI: 10.1109/TCYB.2016.2621129
19.
Rubio-LargoA., Vega-RodríguezM.A., and González-ÁlvarezD.L. (2016b). Hybrid multiobjective artificial bee colony for multiple sequence alignment. Appl. Soft Comput., 41, 157–168.
20.
Rubio-LargoA., Vega-RodríguezM.A., and González-ÁlvarezD.L. (2016c). A hybrid multiobjective memetic metaheuristic for multiple sequence alignment. IEEE Trans. Evol. Comput., 20, 499–514.
21.
SieversF., WilmA., DineenD., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol., 7, 539–539.
22.
YamadaK.D., TomiiK., and KatohK. (2016). Application of the MAFFT sequence alignment program to large data reexamination of the usefulness of chained guide trees. Bioinformatics, 32, 3246–3251.