
Abstract
Abstract
1. Introduction
A
A genome-wide ChIP study produces thousands or more DNA fragments consisting of several hundred base pairs, which cover the binding sites for a TF (Ikebata and Yoshida, 2015). Providing a set of DNA fragment sequences associated with a TF, by comparing the motifs discovered from these sequences with the known TF-binding motifs in a TFBSs database, for example, JASPAR (Sandelin et al., 2004), TRANSFAC (Wingender et al., 2000), we can not only recognize the binding sites for the ChIPed TF but also recognize the cofactor motifs that regulate the TF activity (Smith et al., 2005; Bailey, 2011; Goi et al., 2013).
The majority of early motif-finding algorithms can be divided into two categories. One is word-based (based on the string) methods that mostly adopt exhaustive enumeration, such as Weeder (Pavesi et al., 2001); the other is probabilistic-based (model-based) algorithms, for instance, MEME [multiple EM (expectation maximization) for motif elicitation] (Bailey and Elkan, 1994), AlignACE (Hughes et al., 2000), and ANN-Spec (Workman and Stormo, 2000). The word-based enumerative methods ensure global optimality, and they are appropriate for short motifs finding. Therefore, they are frequently used for motif finding in eukaryotic genomes where motifs are usually shorter than prokaryotes (Das and Dai, 2007). However, the methods cited earlier are not suitable for handling ChIP-seq data. Some of them have undergone reconstruction, arising from many ChIP-tailored algorithms. STEME (suffix free EM for motif elicitation) (Reid and Wernisch, 2011), a ChIP-tailored version of MEME, utilizes a branch-and-bound technology to remove negligible oligomers with significantly low probabilities effectively. For the sake of reducing the computational load in the counting operation, DREME (motif discovery in transcription factor—ChIP-seq data) (Bailey, 2011) and CisFinder (Sharov and Ko, 2009) adopt similar strategies, taking the risk of missing vital motifs in earlier steps of the recursion. Although Hegma (Ichinose et al., 2012) is competitive among current algorithms, the degradation of its detection accuracy is nonignorable.
The model-based approaches are mostly based on the EM (expectation maximization) algorithm (Bailey and Elkan, 1994) or Gibbs sampling (Lawrence et al., 1993). The RPMCMC (repulsive parallel MCMC algorithm for discovering diverse motifs) algorithm is a parallel version of the commonly used Gibbs sampling, running on a parallel-interacting Gibbs sampler. When the routes of different sampling chains are close to each other, a repulsive force (defined as a function of position probability matrices [PPMs]) will drag them forward in different directions. As a result, different sampling chains are urged to seek various regions so that the RPMCMC is more likely than old methods to discover much more diverse motifs. For old and other recent new motif discovery methods, we make a summary that they commonly exhibit low detection accuracy, are instable, and are time consuming. Thus, we are devoted to overcoming these drawbacks and improving them. As with the RPMCMC algorithm, we use parallel mutually exclusive Gibbs sampling to obtain the initial motif. When another motif similarity measure method and motif clustering method are applied to motif screening, we achieve the same or even better performance as the RPMCMC algorithm. These two methods will be demonstrated later.
The main purpose of this study is to derive a novel motif discovery algorithm, which acquires higher detection accuracy while holding competitive computational efficiency. Moreover, the novel method can detect much more believable diverse motifs. To cater to these needs, we propose a new motif-finding algorithm known as GSMC, based on Parallel Gibbs sampling (Lawrence et al., 1993) and Maximal Cliques (Zhang and Chen, 2016) clustering. We adopt parallel-interacting Gibbs sampling for generating initial motifs and then we utilize Maximal Cliques to group initial motifs into different clusters according to Similarity with Position Information Contents (SPIC) (Zhang et al., 2013) among motifs. Finally, we consider the first motif in each cluster as an output motif. On the basis of the ZOOPS (Zero or one motif occurrences per dataset sequence) sequence model, we design the GSMC algorithm without the OOPS constraint and motif length limit (Yu et al., 2015). We implement the GSMC algorithm with C++. When a set of TF ChIP-seq datasets of the ENCODE project (Dunham et al., 2012) is used to compare the performance of GSMC and two other high-performance algorithms, the results show that GSMC happens to make up for the other two algorithms on 20 ChIP-seq data sets for motif discovering; therefore, GSMC is a practical and alternative method in the field of motif finding.
2. Materials and Methods
2.1. Gibbs sampling model
The parallel Gibbs sampling algorithm applies several parallel-interacting Gibbs samplers to produce PPMs. Here, we provide an overview of the Gibbs sampling model.
Let
Initialization of the algorithm is to choose random starting positions in all sequences and to write them into starting positions set
Step one: predictive update step. One of the N sequences, Si is selected randomly or sequentially. The pattern description
Let
where B is the sum of the bj. The qj is calculated analogously, with the corresponding counts taken over all nonpattern positions.
Step two: sampling step. In the sequence Si, each segment of width W is considered a possible instance of the pattern.
After normalization,
2.2. GSMC algorithm
The GSMC algorithm is composed of two parts, which include motif generating and postprocessing. GSMC shares some similarities with RPMCMC, especially in the phase of motif generating. First, we utilize parallel-interacting Gibbs sampling to produce various motifs. Second, based on SPIC and information content (IC), we employ maximal clique clustering to group motifs into different categories. Finally, we select the optimal motif in each category as the final output motif.
2.2.1. Motif generation
We use the ZOOPS model (Bailey and Elkan, 1994) in which there is zero or one motif occurring per sequence. As the input sequences,
Provided the input sequences S, the objective is to detect the PPM
As shown in Table 1 and Table 2, motif generation comprised two parts, which include initialization and updating. The stage of updating relies on Gibbs sampling.
PPMs, position probability matrices.
The number of replicas,
2.2.2. Postprocessing: clustering motifs
After accomplishing the section of motif generation, we can attain massive motif PPMs. Nevertheless, most of them are either false or redundant motifs. It is essential to filter and cluster PPMs for picking out true motifs.
First of all, we get rid of many false motifs according to their IC value less than a cutoff value, which is given by Formula (6):
Then, we use the SPIC metric to compute the similarity score between each pair of rest motifs. As is shown later, the SPIC metric is given by Formulas (7), (8), (9), and (10). For a motif Mx containing nx sequences with length Lx, let
where
For two motifs M1 and M2 with PSSMs P1 and P2, and PFMs F1 and F2, respectively, the similarity score between position X of M1 and position Y of M2 is defined as
where
Further, we construct a motif similarity graph on the basis of similarity score between each pair of motifs. In the graph, each node stands for a motif. Two nodes will be linked by an edge if and only if the weight value is greater than a preset threshold. The weight value of two connected motifs is the similarity score between them. More specifically, binding site motifs that belong to the same TF are more likely to form highly connected sub-graphs with high edge weights in the motif similarity graph than are those from different TFs or spurious motifs. Therefore, if one motif has a low similarity score with any additional motifs, there is a strong possibility that this motif is spurious. These motifs will be deleted from the graph.
Finally, we utilize Maximal Cliques to cluster motifs on the basis of the motif similarity graph. Clustering motifs via Maximal Cliques consists of four steps. A brief introduction of the four steps is given next.
Step 1: For each node, find a maximal clique associated with it, as depicted in Figure 1. First, sort the neighbor nodes of v in ascending order by the weights of their edges incident to v to get an array
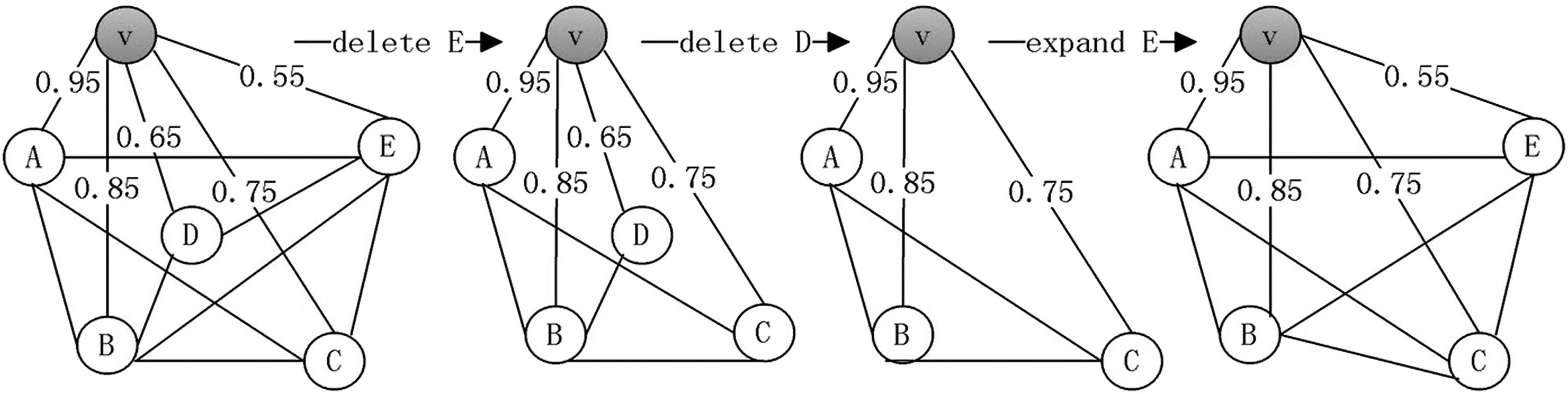
An example of finding maximal clique associated with node v.
Step 2: Merge cliques into clusters. Initially, sort all the unassigned cliques in descending order by the sum of edge weights. Then, compare the first clique with any other unassigned clique (Ci and Cj), if the overlap ratio of the nodes in Ci appearing in Cj divided by Ci is no less than a preset threshold
Step 3: Delete redundant nodes. Compare the latter cliques with the former, and remove the nodes in the latter cliques appearing in the fore.
Step 4: Sort clusters. Sort all the clusters in a decreasing order of edge-weight sum. Choose the first motif in each cluster as the final output motif.
2.3. Performance assessment
We report the performance of three motif discovery algorithms on the dataset: 20 TF ChIP-seq datasets of the ENCODE project. ChIP-seq experiments produce a great amount of DNA segments, which contain many TFBSs bound to a certain TF. In addition to finding these TFBSs, we can discover some cofactor motifs that are involved in the regulatory module of the primary TF. The performance evaluation of the algorithm is outlined next.
First of all, to compare motif detection accuracy among three algorithms, each predicted motif (PPM) by them is matched to JASPAR CORE motifs by using the online TOMTOM tool (Gupta et al., 2007). For a given predicted PPM, TOMTOM outputs the matching scores to all annotated TFBSs (the name of TFBSs) in JASPAR with the statistical significance (E-values: the expected number of false positives in the matches). For each algorithm, a diversity of the discovered motifs is evaluated with the number of known motifs in JASPAR CORE that are matched significantly to the produced PPMs with the acceptable level of significance at E-value less than 0.05 (Ikebata and Yoshida, 2015). The less the E-value, the higher the detection accuracy. For advanced options of TOMTOM program, we choose Pearson correlation coefficient as the motif column comparison function and set the significance threshold at 0.05. Then, we compare the total running time between the RPMCMC algorithm and our GSMC algorithm. Finally, we make a comparison of the number of cofactor motifs among three algorithms.
2.4. Programs and parameter selection
The RPMCMC program used in the article was released on January 6, 2015 (http://daweb.ism.ac.jp/yoshidalab/motif). The DREME program is available online (http://meme-suite.org/tools/dreme). Two motif discovery algorithms (RPMCMC and GSMC) were compiled and installed on 64-bit Ubuntu 14.04 in VMware. The DREME program is directly utilized online. To ensure a comparison that is as fair as possible among the three motif discovery algorithms, the values of the adjustable parameters in RPMCMC and GSMC were selected so as to obtain the optimal performance respectively. For the RPMCMC algorithm, we keep a default value of some parameters and change the others. We modify the number of iterations to 70 and the number of replicas to 20. For the DREME algorithm, we keep the value of all parameters unchanged because it is available online from the site http://meme-suite.org/tools/dreme. For the GSMC algorithm, the number of iterations and replicas are the same as the RPMCMC's. As a section of the GSMC program, clustering motifs are involved in several parameters, such as motif similarity cutoff, IC threshold,
DREME; GSMC, Combining Parallel Gibbs Sampling with Maximal Cliques for hunting DNA Motif; IC, information content; RPMCMC.
3. Results and Discussion
3.1. Parameter optimization and performance comparison
Empirically, we set the value of the similarity cutoff parameter to 0.6 and the values of parameters
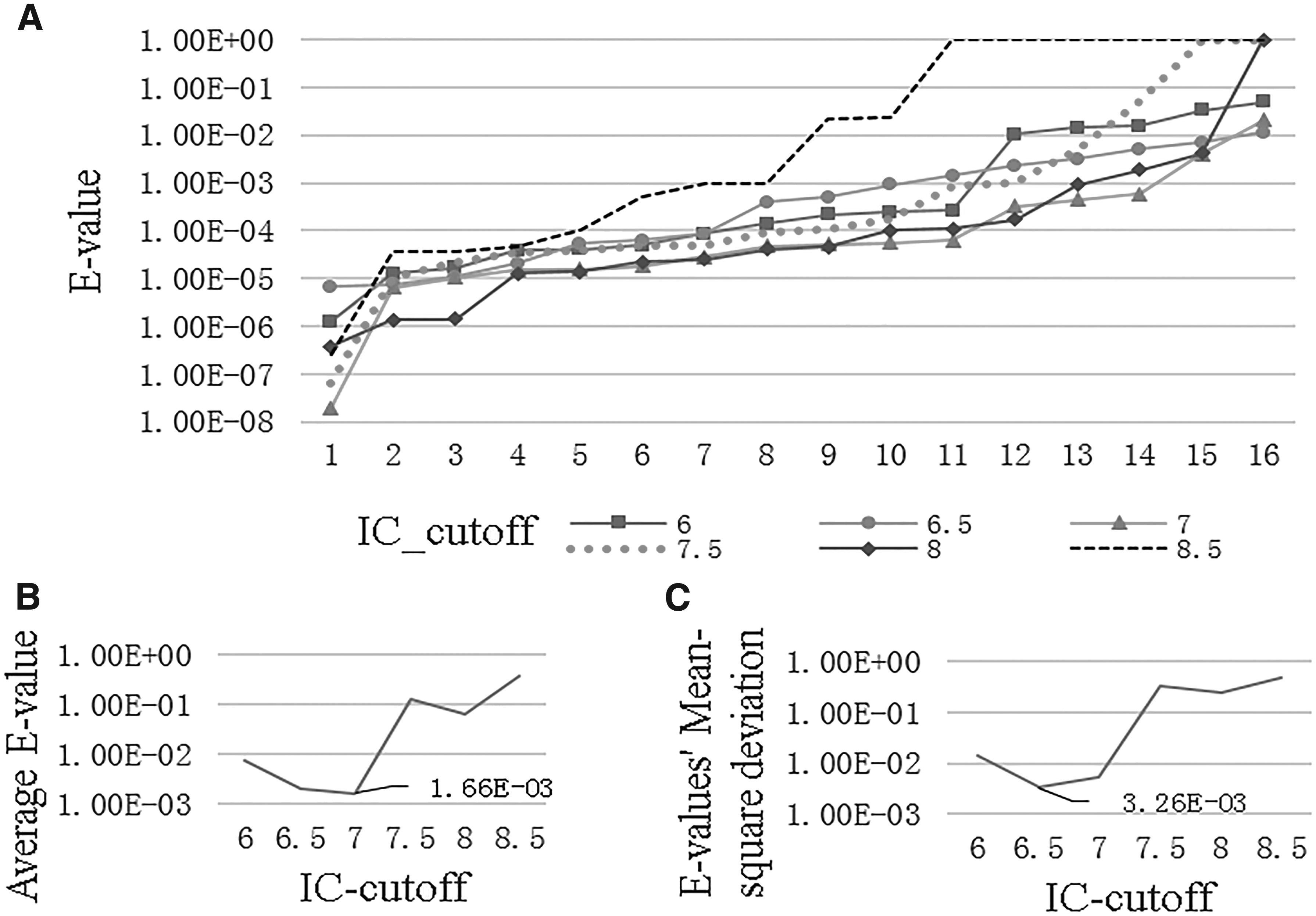
Optimal value selection of the parameter IC threshold.
According to Figure 2B, we can refer to the relationship between the average detection accuracy and the IC threshold value. The less the average E-value, the higher the average detection accuracy. From Figure 2C, we know the fluctuations of the 16 minimal E-values. The less the E-values' mean-square deviation, the higher the stability. Above all, 7 is chosen as the optimal value of the parameter IC threshold in that there is the average least E-value and the second least mean-square deviation in this case.
3.2. Performance on discovering motifs for ENCODE ChIP-Seq datasets
We use the same ENCODE datasets as the paper (Ikebata and Yoshida, 2015). They are SYDH TFBS narrowPeak files (available from NCBI's Gene Expression Omnibus using the accession number GSE31477), and we can download them from http://hgdownload.cse.ucsc.edu goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs/. We choose 20 datasets with different sizes from the 228 ENCODE ChIP-seq datasets as our experimental data. GSMC is implemented in C++, RPMCMC is available on the authors' websites, and DREME can be used online directly. All the tests are conducted on Intel® Core™ i3 processor with 4-core CPUs and 4 GB of main memory. On the virtual platform of Ubuntu 14.04, we run GSMC and RPMCMC for 20 datasets, record running time, and save all the produced PPMs. With the help of the TOMTOM, we compare the produced PPMs with all the known motifs in JASPAR and leave the least E-value of matching between the ChIPed motif and the query motifs. Then, we make comparisons among GSMC, RPMCMC, and DREME in terms of detection accuracy, computational speeds, and the number of cofactor motifs discovered. From a performance perspective, our GSMC algorithm makes up for rather than takes the place of existing DNA motif-finding methods for analyzing the large ChIP-seq data.
3.2.1. Detection accuracy
Each dataset corresponds to a ChIPed TF, which has a relevant motif. We run each dataset for 50 times and each time, we make a note of the least E-value of all meeting matches (E-value <0.05) that are aimed at the ChIPed motif. Figure 3 illustrates the comparison of detection accuracy among three algorithms on a ChIP dataset (wgEncodeSydhTfbsHepg2Nrf1IggrabPk) in which the binding sites of NRF1 were studied in cell HepG2. When it comes to detection accuracy, DREME surpasses the others and GSMC is nearly equal to RPMCMC.
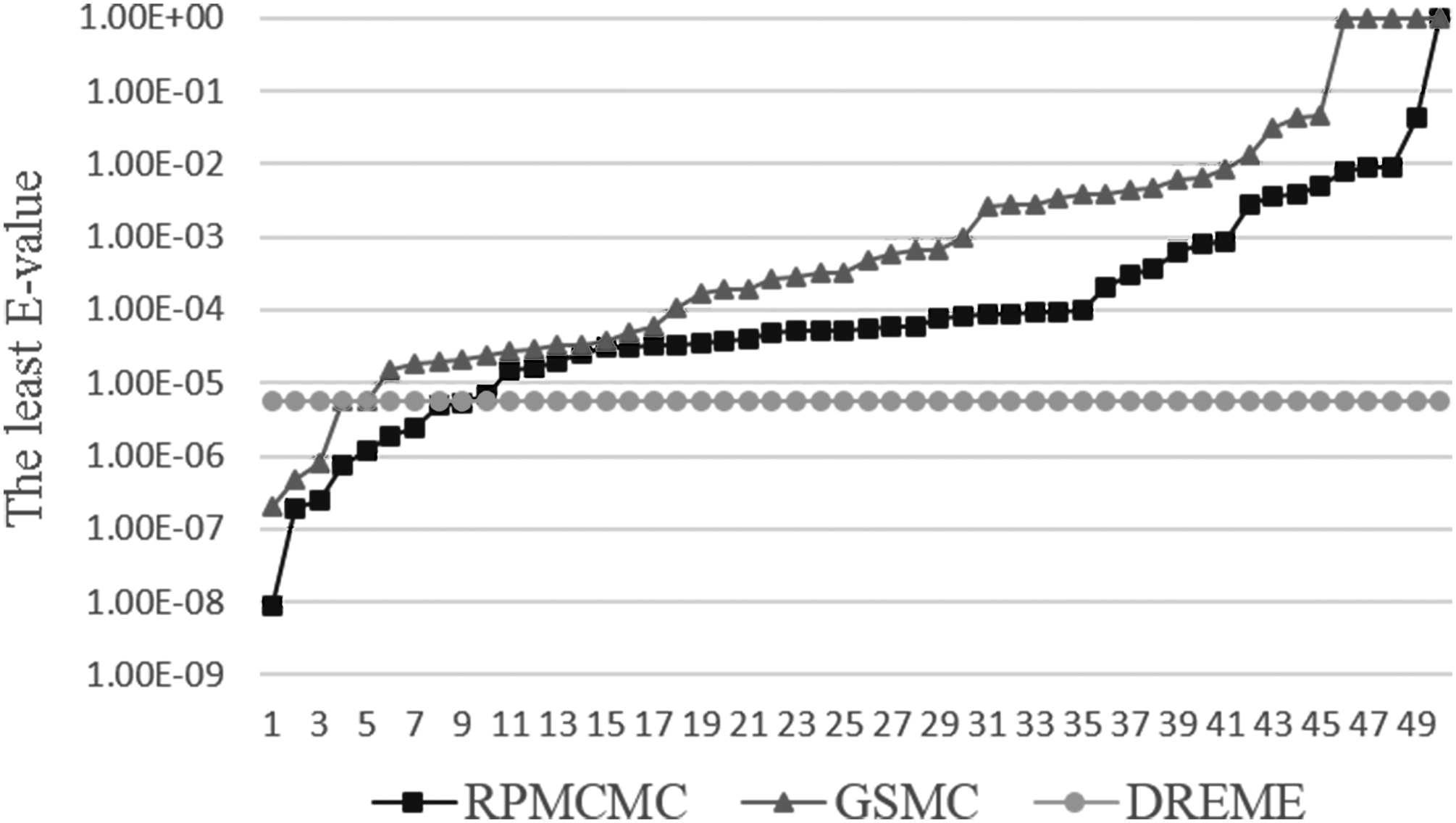
Comparison of motifs' detection accuracy among RPMCMC, GSMC, and DREME on datasets (wgEncodeSydhTfbsHepg2Nrf1IggrabPk). DREME; GSMC, Combining Parallel Gibbs Sampling with Maximal Cliques for hunting DNA Motif; RPMCMC.
In Figure 3, we have made a comparison of detection accuracy among three algorithms on one dataset. Next, we make comparisons on all chosen datasets. Figure 4 provides the comparison of GSMC with RPMCMC and DREME on 20 chosen datasets. For most datasets, GSMC and RPMCMC are superior in detection accuracy to DREME.
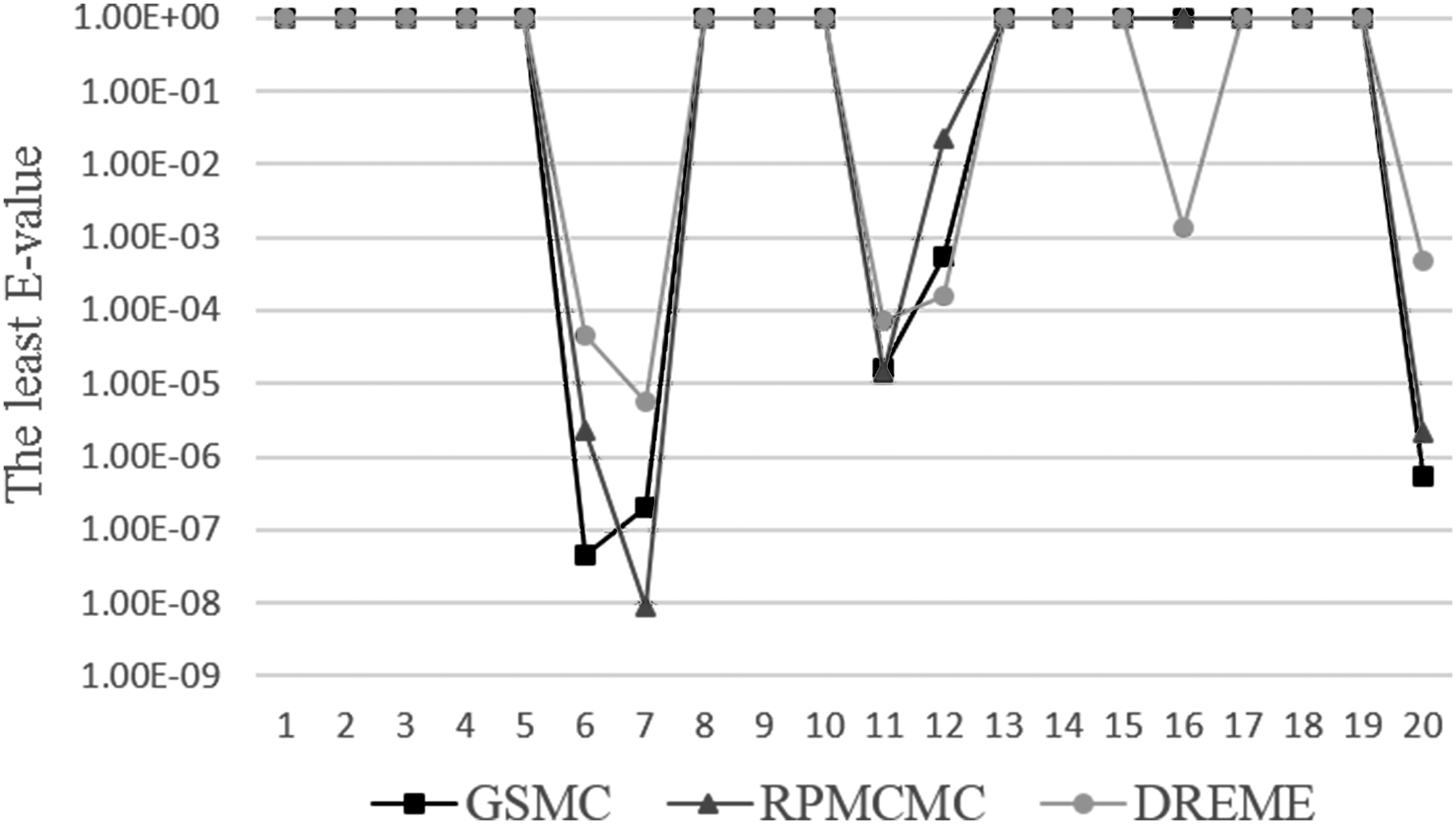
Comparison of motifs' detection accuracy among RPMCMC, GSMC, and DREME on 20 chosen datasets.
3.2.2. Computational speeds
Figure 5 displays the computational time for GSMC and RPMCMC on 20 datasets. Each dataset is tested for 16 times, and we take the arithmetic mean as the running time. In terms of computation efficiency, GSMC is comparable to RPMCMC. Motif producing of GSMC basically conforms to RPMCMC. Therefore, the bottleneck calculating the posterior probabilities of the motif start sites ui in RPMCMC still exists in GSMC. However, the clustering section of GSMC is superior to RPMCMC in terms of flexibility and scalability. With these performances, GSMC is more likely to be a high-efficient method in future studies. Though GSMC cannot surpass RPMCMC in computational efficiency, it is a competitive algorithm in the case of computing time.
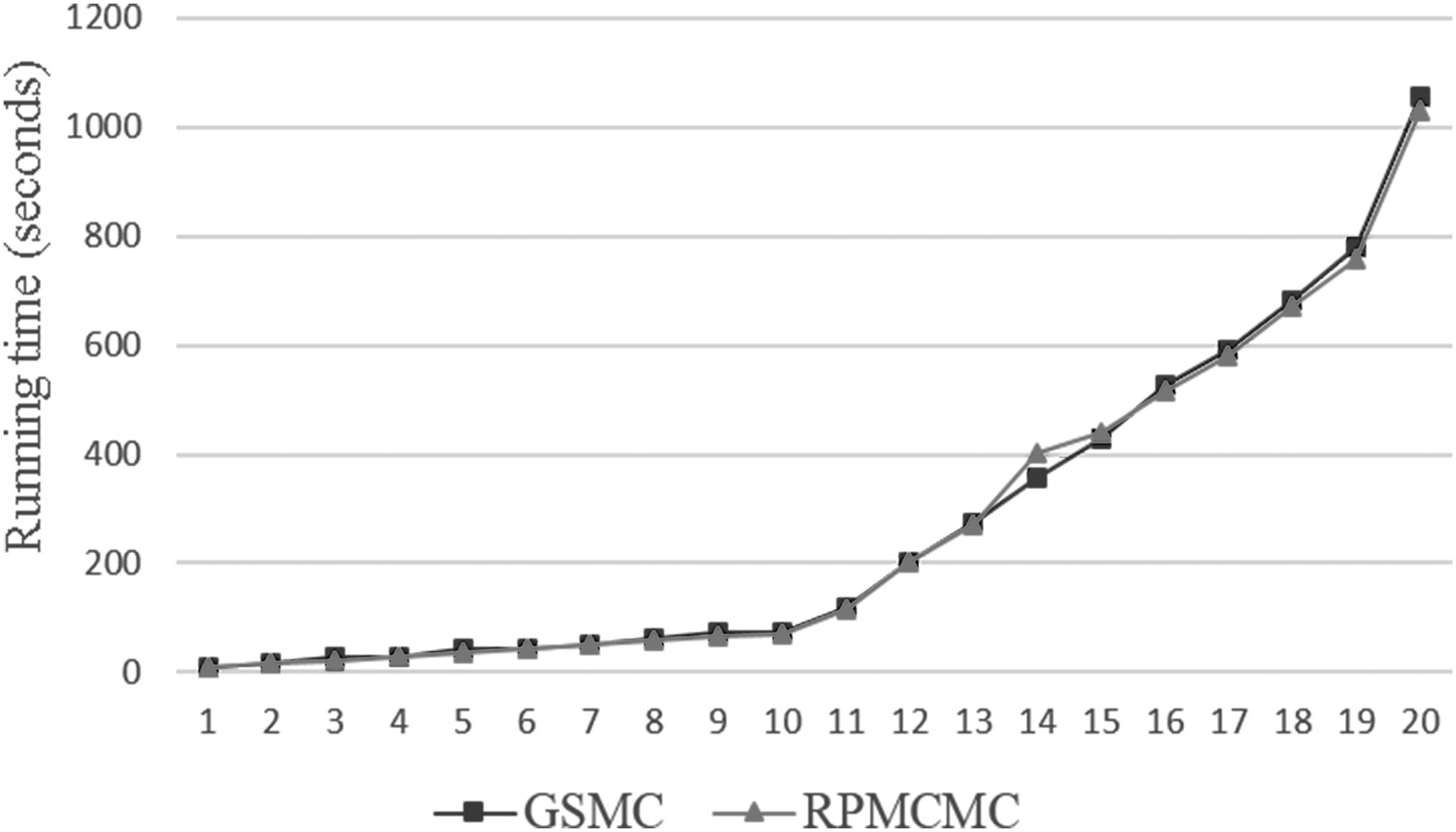
Comparison of computational efficiency between GSMC and RPMCMC. Running time is the CPU execution time.
3.2.3. Comparison on the number of cofactor motifs discovered
A recent study by Chin Lui Goi and Peter Little called cell-type and TF-specific enrichment of transcriptional cofactor motifs in ENCODE ChIP-seq data claims that TF-specific interactions between TFs and cofactors are essential for transcriptional regulation through recruitment of general transcription machinery to gene promoter regions (Goi et al., 2013). It is also mentioned in the study that some of the cofactor motifs are experimentally verified cofactors and others are potentially novel cofactors. The same can be said of the cofactors discovered in our study. Even with all that, they have evident reference value and directive significance to further studies. Therefore, the ability of detecting cofactor motifs is crucial to a motif-discovering algorithm. Just as shown in Figure 6, the cofactor motifs found by GSMC outnumber those found by RPMCMC on almost all datasets.
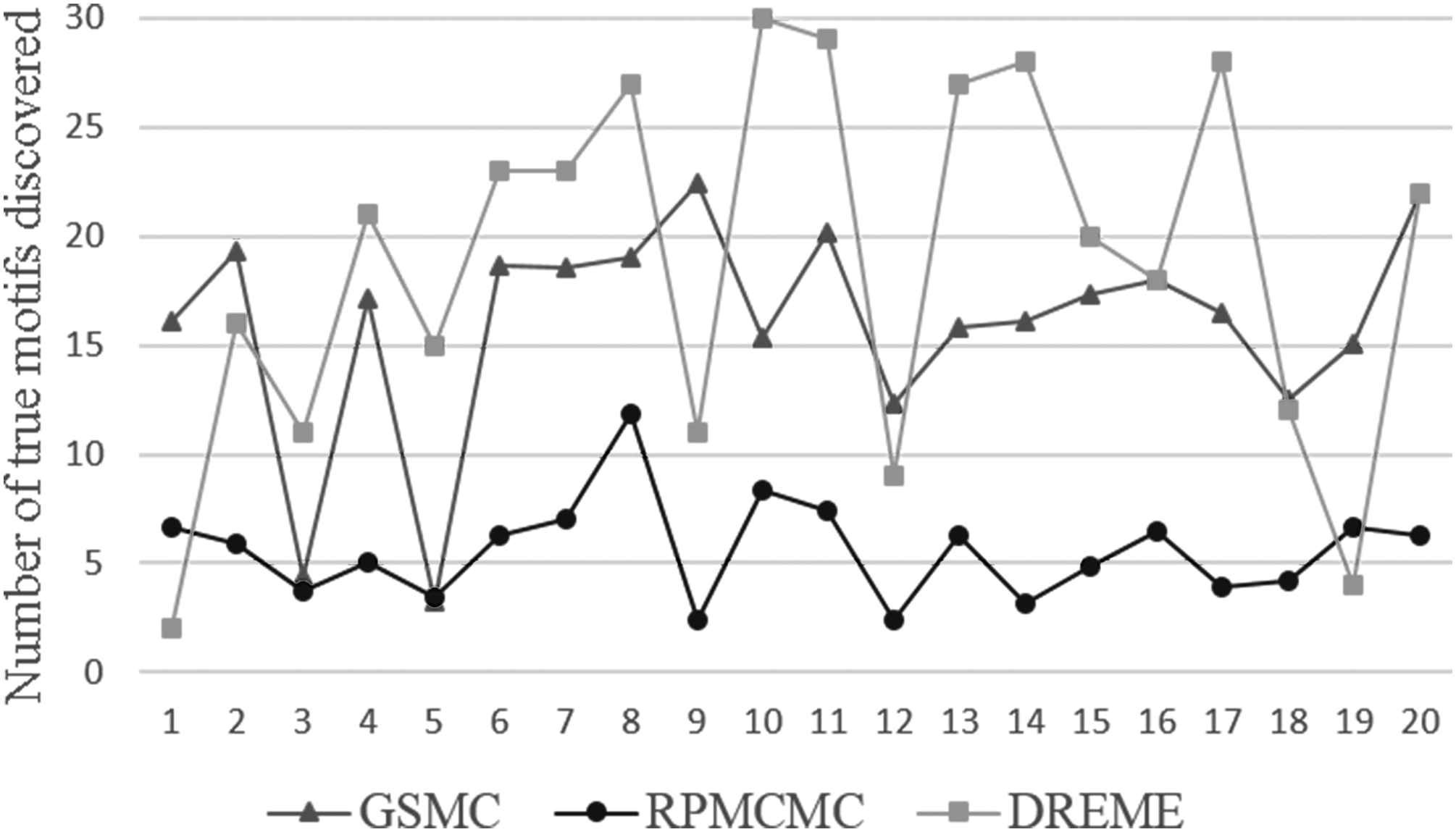
Comparison on the number of cofactor motifs. Each dataset is tested 16 times, and each data point in the figure is the mean value of 16 tests.
Obviously, GSMC is superior in the number of cofactor motifs to RPMCMC. In other words, the ability of GSMC to detect cofactor motifs is better than RPMCMC and basically equal to DREME.
4. Conclusion
Many older popular motif finders cannot be applied to handling large datasets generated by ChIP-seq (Reid and Wernisch, 2014). Though a vast amount of novel algorithms spring up for handling huge datasets, most of them attend to computation speed and lose sight of the accuracy of motif detection. To overcome the drawbacks of them, we develop a ChIP-tailored motif discovery tool called GSMC, which hunts for DNA motifs by combining Parallel Gibbs Sampling with Maximal Cliques. In terms of computation time and detection accuracy, GSMC can rival the most recent motif discoverer RPMCMC, which was specifically designed to handle large datasets. Moreover, GSMC is capable of detecting much more known and potential cofactor motifs where GSMC far exceeds RPMCMC. Besides, GSMC is superior in accuracy of motif detection to DREME. On the whole, we present a novel motif finder reconciling computation speed with detection ability. Locating these motifs plays a critical role in identifying transcriptional regulation. We expect that GSMC will have a place in the process of exploring gene expression.
Footnotes
Acknowledgments
This work was supported by the grants of the National Science Foundation of China (Grant Nos. 61472467, 61672011, and 61471169) and the Collaboration and Innovation Center for Digital Chinese Medicine of 2011 Project of Colleges and Universities in Hunan Province.
Author Disclosure Statement
No competing financial interests exist.