
Abstract
Abstract
MetReS (Metabolic Reconstruction Server) is a genomic database that is shared between two software applications that address important biological problems. Biblio-MetReS is a data-mining tool that enables the reconstruction of molecular networks based on automated text-mining analysis of published scientific literature. Homol-MetReS allows functional (re)annotation of proteomes, to properly identify both the individual proteins involved in the processes of interest and their function. The main goal of this work was to identify the areas where the performance of the MetReS database performance could be improved and to test whether this improvement would scale to larger datasets and more complex types of analysis. The study was started with a relational database, MySQL, which is the current database server used by the applications. We also tested the performance of an alternative data-handling framework, Apache Hadoop. Hadoop is currently used for large-scale data processing. We found that this data handling framework is likely to greatly improve the efficiency of the MetReS applications as the dataset and the processing needs increase by several orders of magnitude, as expected to happen in the near future.
1. INTRODUCTION
T
MetReS was built by matching the KEGG gene names to their NCBI names and synonyms. The database includes full gene names and synonym tables for ∼10,000 organisms with fully sequenced genomes. The database was implemented by using MySQL technology.
Biblio-MetReS is a user-friendly tool for the automated reconstruction of literature gene/protein networks implemented in Java, which combines on-the-fly analyses of the full text of scientific documents that are freely available on the Internet with preprocessed documents that were found in previous searches. Although the on-the-fly approach has high run times, it keeps the information available to the user as up to date as possible. To circumvent this high runtime, other tools use only preprocessing of the documents, Barbosa-Silva et al. (2010), Hoffmann and Valencia (2005), Szklarczyk et al. (2011) at the cost of providing information that is somewhat out of date. Preprocessing of texts/documents consists of analyzing the documents offline, extracting the relevant information, and creating an adequate table to store that information inside the database. Combining this strategy with high performance during database access leads to a high level of Quality of Service (QoS). However, parallel access to the document database can cause bottlenecks, thus being a major concern that needs to be dealt with.
Homol-MetReS is a web application that allows simultaneous large-scale re-annotation, functional integration, and automatic comparisons of metabolic networks on a multiple full genome scale. It automates comparisons that would otherwise be done semi-manually, for example, using PathBlast (Kelley et al., 2004), KEGG (Chaudhury and Igoshin, 2009), or MetaCyc (Veening et al., 2008). The functionalities in Homol-MetReS that have the highest needs for processing power and database access optimization are the comparison between fully sequenced genomes and the classification of large numbers of homologous proteins according to specific biological process categories. As in the case of Biblio-MetReS, database access is a bottleneck in server runtime. Our interest is focused on expanding this centralized database into a distributed one by using additional resources located in a cloud infrastructure.
In Lin et al. (2009), the authors dealt with the challenge of optimizing a MySQL database for novelty detection, which is the process of singling out novel information from a given set of text documents to optimize the database tables for up to 10 million records. In Bujdei et al. (2008), the authors also used database optimization and SQL tuning to achieve better performance levels for health monitoring systems. However, there is no work determining the boundaries of tolerable SQL performance and even less for specific databases of genomic data. The requirement to perform compute-intensive analytics on (semi) structured bulk datasets (as in our case) has pushed SQL-like centralized databases to their limits (Abadi, 2009). This fact, along with the highly parallel nature of these tasks, has led to the development of horizontal scalable, distributed nonrelational data stores, called NoSQL systems, which are widely used in big data and real-time web applications. Google's Bigtable (Chang et al., 2008), Amazon's Dynamo (DeCandia et al., 2007), Facebook's Cassandra (Lakshman and Malik, 2010), LinkedIn's Voldermort, and Apache's Hadoop (Lam, 2010) are examples of the most important NoSQL systems.
In this article, two architectures for a genetic database (MetReS) are presented, one following an SQL design and another with an NoSQL system implemented with Apache Hadoop. * Hadoop can be a useful solution among many open source solutions for large-scale storage and processing of datasets, since it stores the data in Hadoop Distributed File System (HDFS) and provides the platform that can process them with the simple MapReduce programming model (Dean and Ghemawat, 2008). For this reason, Hadoop is well suited to large dataset analysis and its deployment in a cloud-based infrastructure.
2. METHODS
This section describes the architectural design of the cloud framework, where its main features are presented and analyzed. Then, the two biological tools are thoroughly described. Finally, the database shared between the two biological applications is summarized.
2.1. Cloud architecture
As stated in Xiong and Perros (2009) and discussed earlier in Section 1, most current cloud computing infrastructures consist of services that are offered and delivered through a service center that can be accessed from a web browser anywhere in the world. The front-end and the back-end are the two most significant components of a cloud-computing architecture. Their main features are presented later.
The front-end serves the web requests or tasks arriving from the applications. In Biblio-MetReS, a desktop application acts as the front-end together with a web service. In the case of Homol-MetReS, the front-end is the web browser itself. In both cases, the front-end sends the requests to the web server, which is also in charge of scheduling, controlling, and executing the tasks in the back-end. Tasks delivering requests to the cloud service are then associated with an SLA (Service-Level Agreement). Our work is focused on guaranteeing the SLA, which is mainly done in the back-end. The interface and negotiation processes are usually performed at the front-end. In our case, we did not implement any infrastructure supporting SLA negotiation, as it is not yet needed. However, as the number of users and the database size of the applications increase, such an infrastructure will be implemented.
The back-end functions include managing the job queue, as well as the processing and storage servers. The processing servers are responsible for performing most of the computation. They are also responsible for sending service requests to the storage servers, which, in turn, manage the database. We designed our back-end to support multiple processing servers, so multiple concurrent requests can be serviced, and, consequently, concurrent requests to the database can be delivered at the same time to guarantee SLA (Vilaplana et al., 2013). In doing so, we propose some solutions to provide a high level of QoS for the analysis of large data sets, thus determining a means to fix the SLA for accessing the database for the particular case of our subject applications. Biblio- and Homol-MetReS are two applications with high I/O requirements, and, consequently, QoS is mainly focused in the Internet connection and the database. On this occasion, our research was focused on the database performance. This is justified because database size is growing faster than all other factors. Although the initial database contained ∼700 genomes in 2011, it grew to 1100 genomes in 2012, 1500 genomes in 2013, and 10,000 this year. The sharp increase between 2013 and 2014 is also due to the fact that viruses are now also included in the database.
A cloud architecture composed of virtual machines implemented using the open-source OpenStack framework was designed. One virtual machine hosted the web server. We designed two different database architectures attached to the cloud hosting the Biblio- and Homol-MetReS database. These architectures depend on the database implementation. The first one was implemented by using a standard relational MySQL database embedded in one OpenStack virtual machine. This is the most traditional and widely used architecture. We then designed an NoSQL system by using the Apache Hadoop framework implemented as a cluster platform made up of a cluster system.
Job response time is one of the most important QoS metrics in a cloud-computing context (Aversa et al., 2011). For this reason, it was also selected as the main QoS parameter for evaluating the architecture performance. Nah (2003) stated that for users, the tolerable response time before abandoning a web page varies with different circumstances and contexts. However, the findings from this study suggested that most users are only willing to wait for about 2 seconds for simple information retrieval tasks on the web. So, if this result is applied to cloud computing, a reasonable design decision should be to design cloud systems with QoS (i.e., response time) as low as possible. Designing an efficient database system, depending on the database accesses, can be a challenge.
2.2. Biological tools
There are important tools in the literature for the automatic identification of co-occurrence of genes/proteins from the Internet to assist in the reconstruction of molecular circuits, such as Laitor (Barbosa-Silva et al., 2010), iHOP (Hoffmann and Valencia, 2005), String (Szklarczyk et al., 2011), and Biblio-MetReS. Of these, Biblio-MetReS is the only tool that is able to search for gene co-occurrences in sentences, paragraphs, and the overall document on the fly. This allows Biblio-MetReS to more fully assess the interactions between a pair of genes.
In the Biblio-MetReS application, a desktop application acts as a client of the web service. In the case of Homol-MetReS, the client is the web browser itself. Requests delivered to the data center are associated with an SLA. Our work is focused on guaranteeing the SLA. On this occasion, the database performance was the main focus of our research. In doing so, we propose some solutions to provide a high level of QoS, thus determining a means to more efficiently implement the SLA in accessing the database for the particular case of our subject applications.
The use of model organisms for research is a hallmark of scientific endeavor (Jin et al., 2009). The accumulation of fully sequenced genomes (Siva, 2008) and the advances in comparative genomics (Ellegren, 2008; Tettelin et al., 2008) and computational systems biology (Alves et al., 2008) allow us to develop strategies that compare the protein or gene networks involved in the process of interest to establish similarities. These similarities can be used to predict, to a first approximation, the accuracy of extrapolating the behavior of specific processes between organisms. Testing this idea requires a thorough analysis of the molecular circuits in a well-known model organism and a comparison of these circuits to those in other living beings. In fact, a gap exists in systematically establishing how close different organisms are with respect to a given process, before choosing one of them as a model for studying that process (Karathia et al., 2011). Provided the MetReS database of fully sequenced genomes, Homol-MetReS implements innovative functionality in such a research field. Thus, as in the case of Biblio-MetReS, we are interested in providing Homol-MetReS with SLA guarantees.
2.2.1. Biblio-MetReS
Biblio-MetReS is a Java client-server application that analyzes co-occurrence of genes/proteins and biological functions in online documents. To do so, it needs a database to efficiently mine the documents and manage information about organisms, genes/proteins, the processes in which the genes are involved and the relationships between all these entities. Biblio-MetReS searches the Internet for documents with data-mining techniques and operates as follows. Users must register to log in in to Biblio-MetReS. After login, users must choose an organism to work with. The application loads all the genes for the selected organism in the database. Once this is loaded, the user is presented with the main window (Fig. 1), where she/he can select data sources as well as genes to search for in those data sources across the Internet. The data sources are general search engines (Yahoo, Live Search, Ask, Answers, Altheweb, and Lycos) and literature databases (Medline, PubMed, Biomed Central, PLoS, Bentham, Highwire, SCOPUS, and Elsevier).
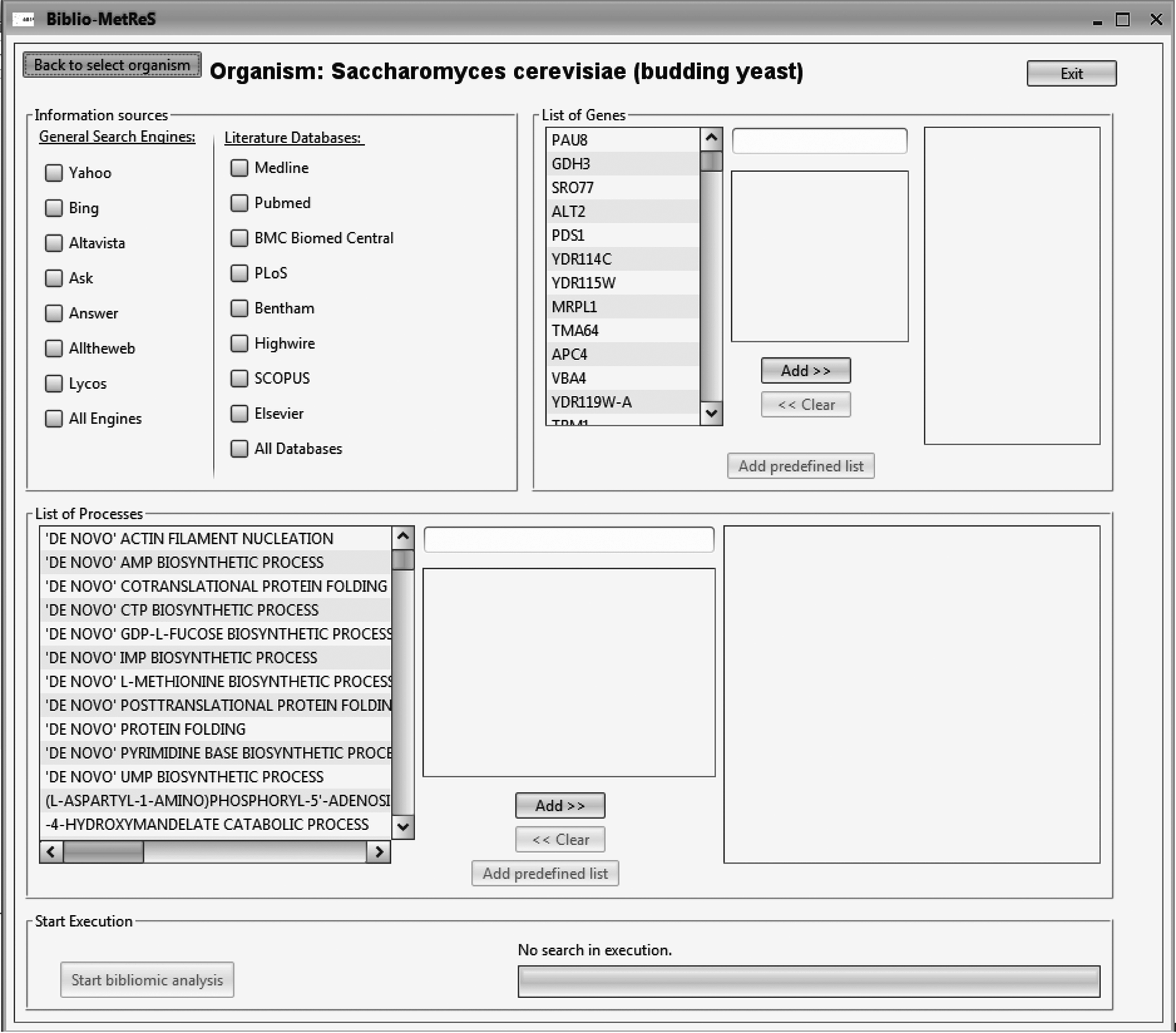
Bilio-MetReS.
Once the choices have been made and the search has started, the tool identifies and downloads the documents from the selected data sources that contain the gene names used in the search query. Each document is then parsed to identify all other genes and gene synonyms (also contained in the database) for the organism of interest that is also mentioned in the documents. A similar workflow is used to analyze biological processes in the documents. Entity co-occurrence patterns are analyzed and represented in several ways. For more details, see Usié et al. (2011, 2014).
2.2.2. Homol-MetReS
Homol-MetReS is a web application that identifies appropriate model organisms in which to study the functionality of a specific biological process or molecular circuit. Then, the tool permits the functional information of such a model organism to be extrapolated to other organisms where that process or circuit might be hard to study directly due to reasons that range from the technical to the ethical.
Homol-MetReS provides a wide array of features. Here, we focused on those requiring access to the database because they are the major cause of bottlenecks.
Users can automatically compare the sequences of the individual proteins from their proteome of interest with the full proteome of ∼10,000 other organisms that have fully sequenced and annotated genomes. Functional information from one organism can be transferred to another by the user, based on sequence homology. The process for doing so is illustrated in Figure 2. One of the bottlenecks in the computational process underlying this functionality is access to the database. This is so because many requests must be sent (in parallel when possible) to the database to compare the full proteome of an arbitrary number of organisms. These comparisons between organisms must be done within a reasonable time. Operations related to this kind of comparison are dealt with in the Results section.
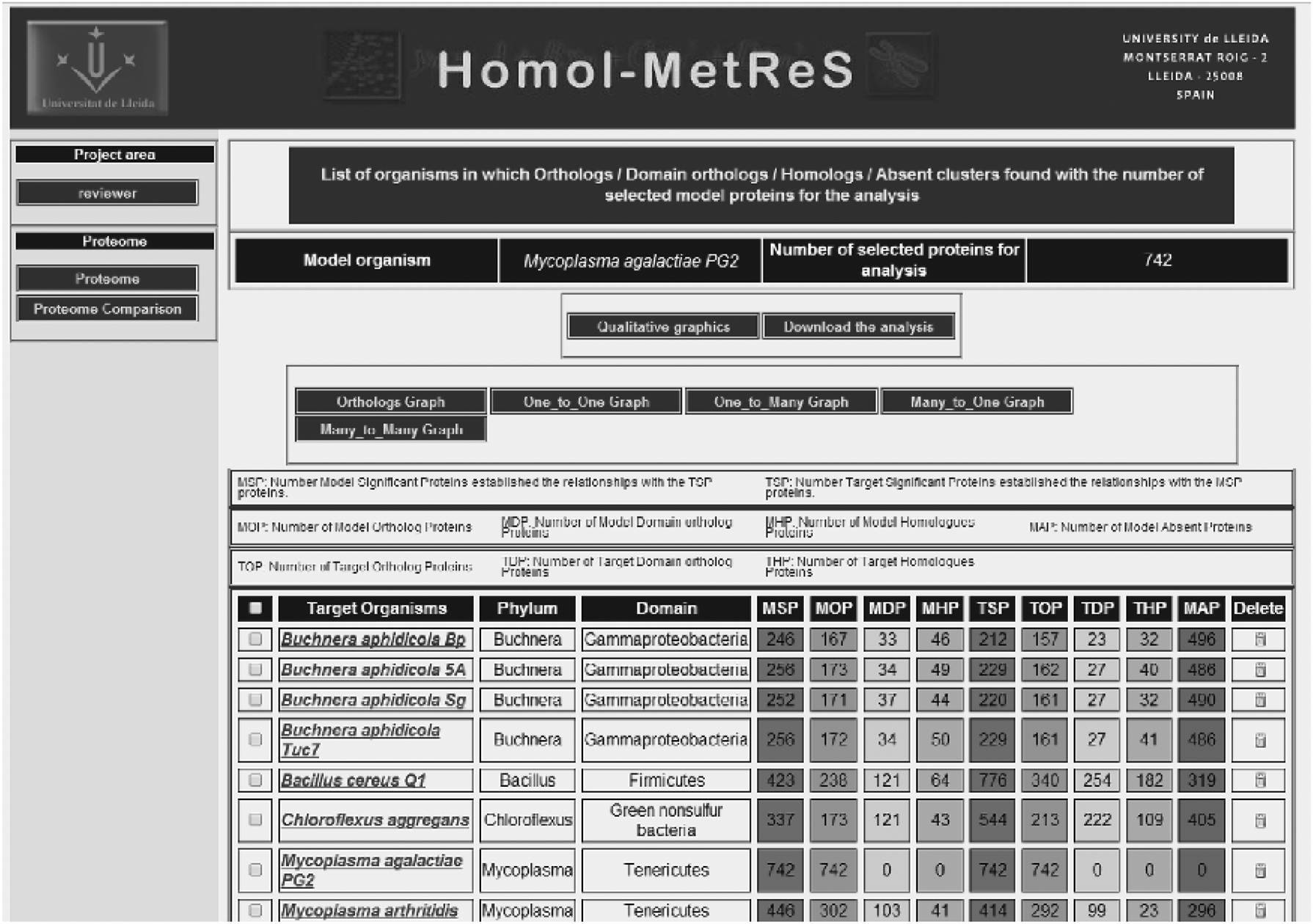
Homol-MetReS.
The database operations used in this functionality can mainly be classified as follows: (a) operations retrieving sequence information for the complete proteomes of any set of organisms contained in the database, (b) operations comparing information data from multiple organisms, and (c) operations to create temporary tables that contain information about the comparative analysis of the proteins among the organisms being analyzed. Third, the tables created in (c) are then used to represent heat maps that enable users to visually compare the similarity between the sets of proteins involved in specific biological processes in all the organisms being analyzed. Neither database access nor I/O is limiting for this functionality. Also note that after a comparison between the proteomes of any two organisms is done for the first time, the results are stored in the database, thus allowing for any future comparisons to be made much faster as only precalculated results need to be looked up, instead of having to recalculate the whole comparison. The downside for this is that an amount of data typically between 2 and 10 times the size of the largest proteome is stored for each comparison.
2.3. Database definition
The shared database used by Biblio-MetReS and Homol-MetReS consists of a collection of tables organized according to the relational model, with defined relationships between each pair of tables. The principal tables correspond to organisms, genes, processes, and documents. These tables contain the information that is most frequently accessed by the applications. The gene's table relates genes, biological processes, and organisms. The document table stores statistical information about the documents found on the Internet by Biblio-MetReS in the data-mining search phase. Our study has focused on the organism and process tables.
Table 1 (Organism) shows an example of one of the tables that contains information about the proteins coded in the genome of a fully sequenced organism contained in the central MySQL database. All such tables follow the same structure, as shown in Table 1, where Org corresponds to the prefix of the organism. The protein sequences, which are strings of characters, are stored in the isoform_sequence field.
Table 2 (Process) shows the description of each process. This table stores information about biological processes associated with genes from one of the organisms with fully sequenced genomes currently included in the central database (e.g., Saccharomyces cerevisiae, Homo sapiens, Escheridia coli K12 MG1655, Drosophila melanogaster, etc.).
Figure 3 shows a box plot representing the mean and quartiles for the relative amino acid usage in all the proteins from all the organisms of the database. These results are consistent with what biologists have known for some time. As an example, we can see in the figures as the amino acids most frequently used in proteins are, on average, A, G, and L. In addition, highly complex and reactive amino acids, such as C, H, or W, are used with very low mean frequencies. This is consistent with the main functions that are known for these amino acids.
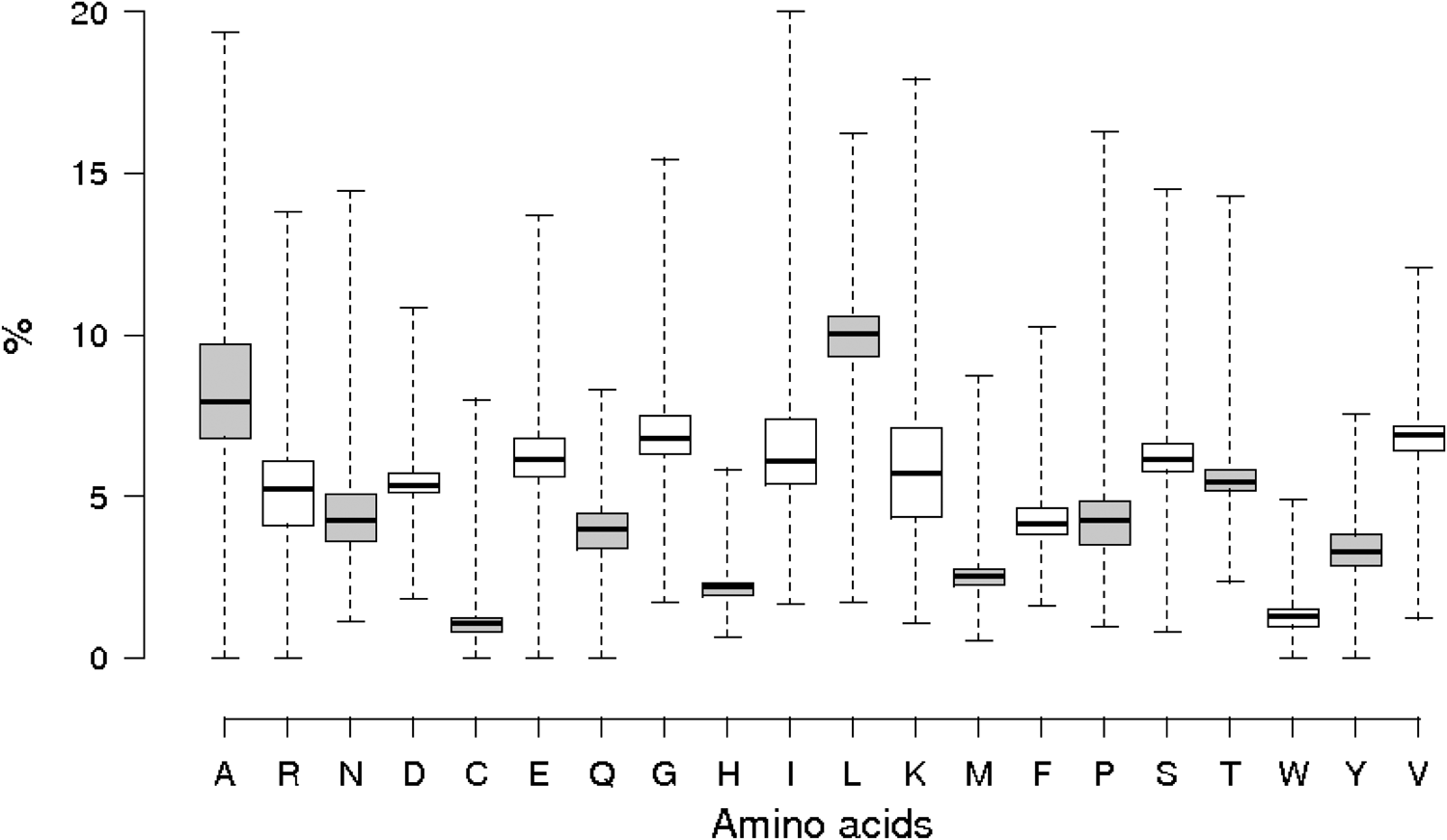
Average relative amino acid frequency in all the organisms. There are 9961 data points.
These results illustrate the capacity of extracting valuable statistical analysis from our current database.
2.3.1. Biblio-MetReS
Incoming documents found on the Internet are individually analyzed by Biblio-MetReS. Once a document has been analyzed, the results are stored in the database by using the tables described earlier, among others. Subsequent searches that identify the same document will retrieve the preprocessed statistics stored in the database, thus avoiding repeating the time-consuming on-the-fly analysis. On average, the database is accessed twice per new document to store statistical information for future use, and five times per document to retrieve the statistical information from preprocessed documents when this information is required in subsequent searches.
2.3.2. Homol-MetReS
In Homol-MetReS, users can perform different kinds of searches that are typically related to which organisms are present in the database or the function of proteins in each organism. For example, these can be for all proteins that are annotated to a specific biological process or have a defined function. These types of query do not impose high demands on computational resources. In contrast, searches that are done to analyze the results of whole proteome comparisons are computationally more demanding. Examples include the search for similar proteins in the various organisms and analysis of the amino acid sequences of those proteins. These analyses can generate relevant biological information about how the complexity of proteomes is correlated to the type of amino acids that are more frequently used in proteins. Because these types of searches use a lot of resources, our analysis is mostly focused on them. Nevertheless, the effect of database architecture on the simpler queries is also analyzed.
3. EXPERIMENTAL RESULTS
As the amount of data grows, a drop in the performance for access to the database shared by Biblio-MetReS and Homol-MetReS is expected. This will lead to an increase in runtime and, therefore, a fall in QoS. Although this performance drop may not be significant for small or simple queries, it could be a severe limitation when working with much larger datasets or when trying to extract more complex statistical analyses from our data. We performed several experiments to assess at what size of the dataset this would happen and if using an Hadoop data handling framework would solve the problem.
These experiments were performed in the two different data-handling systems, MySQL and Hadoop. The main differences between the two frameworks were in the database architecture. The MySQL database server was located in a virtual machine, running on an HP Proliant DL165 G7 node with two Opteron 6274 processors at 2.2 GHz with 16 cores each with 192 GB of DDR3 RAM and a 4.5TB disk. The resources assigned to the virtual machine were 4 cores, 32 GB of RAM and 525 GB of disk. In Hadoop, the original MySQL database server was replaced by a Hadoop framework, made up of a commodity cluster composed of 24 machines with Intel Core 2 Quad at 2.4 GHz with 8 GB of RAM and a 2TB disk, interconnected by a network of 1 Gb bandwidth. Certainly, much more cluster configurations should be considered. However, we chose a compromise with the assert done in Pavlo et al. (2009), where authors confirmed that a MapReduce system (i.e., Hadoop) needs 1000 nodes to match the performance of a 100-node parallel SQL-database system.
Several tests were performed to compare the performance of the MySQL and the Hadoop systems in different scenarios.
3.1. MySQL optimization
First of all, the main running parameters related to optimizing the MySQL database performance were obtained by executing the benchmark MySQLTuner † (Table 3).
The query_cache_size parameter determines the amount of memory allocated to caching query results. The optimal value depends on the system architecture. The key_buffer_size indicates the amount of memory requested. Internally, the server allocates as much memory as possible up to this amount. The query_cache_limit parameter determines the maximum size of cached records.
3.2. Scaling MySQL
The variation in MySQL performance was tested after optimization by varying the computing resources, as described in Table 4. We increase the number of CPUs that the virtual machine could use from 1 to 16 Virtual CPU (VCPU) and the available RAM from 4 to 64 GB. Each Virtual CPU was a QEMU Virtual CPU (cpu64-rhel6) at 2199.998 MHz. A dataset with a total of 10 identical Organism tables with 638,848 entries each was used. The size of each table was 686.55 MB. This dataset has a total size of 6.7 GB.
M, maximum RAM (in GB) allocated; T, execution time (in seconds); V, number of VCPUs.
Figure 4 shows the same test but in terms of efficiency, defined as:
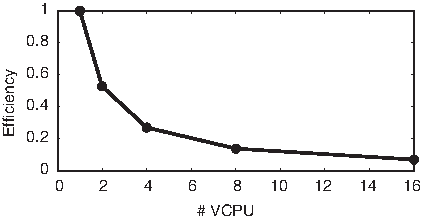
MySQL efficiency.
where S and P represent the serial and parallel execution times, respectively, and N represents the number of VCPUs used. RAM was scaled to be enough for the computation being done. That is, the RAM was never overloaded.
The simplified command to perform this test is shown next:
SELECT t.table_name
FROM information_schema.tables t
WHERE t.table_schema = DATABASE()
AND t.table_name LIKE ‘%t_sequence_comb_%’;
These tests show that, although the MySQL database response time improves by at most a few percent, when the number of VCPUS increases, this comes at the cost of sharply decreasing the efficiency of the process by up to 85%. This justifies the need to look for an NoSQL solution.
3.3. MySQL versus Haddop performance
Two types of tests were performed to quantify the effects of using an optimized regular relational database implemented in MySQL versus the utilization of a Big Data analysis system. First, we compared the performance of the two data-handling frameworks by executing database queries comparable to those regularly done by the users of the Bilio-MetReS and Homol-MetReS applications. In addition, more complex and data-intensive queries were tested.
The performance results achieved by both data-handling systems (MySQL and Hadoop) in both sets of experiments are presented in two separate sections, nondata-intensive and data-intensive queries, in Sections 3.3.1 and 3.3.2, respectively.
3.3.1. Noncomputing-intensive queries
To compare the response time of a commonly used query in data-handling systems, a simple access performance to a subset formed by 24,662 entries of the Process table (called Dataset1) was tested. Dataset1 has a total size of 24.92 MB. Figure 5 shows the total execution time (in seconds) of the following query (load into Memory all the entries in the Process table):
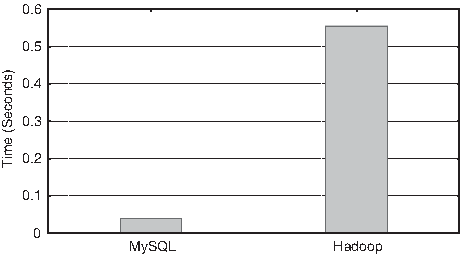
MySQL and Hadoop runtimes when accessing the Process table.
SELECT * FROM Process;
MySQL is one order of magnitude faster than Hadoop in this experiment, although the difference in absolute time between both executions was only 0.515 seconds. Nevertheless, this difference can suppose a dramatic decrease in overall QoS of the web applications if they used Hadoop, because these queries are very common.
A second experiment with a more complex query was carried out. The aim was to obtain the total number of proteins contained in the proteome of each organism. In this case, MySQL and Hadoop used another dataset (called Dataset2), distributed between 9963 tables. The table Organism (defined in Section 2.3) contains 9962 different organisms. Then, one additional table per organism contains the sequences of all proteins coded in the genome of that organism. This dataset has a total size of 14 GB.
The query executed in the MySQL system was implemented by using a Python script described in Algorithm 1. This algorithm counts the number of proteins of all the organisms in the database. An equivalent MapReduce job was implemented for the Hadoop system. Figure 6 shows the execution time (in seconds) of both systems. It can be seen as MySQL was still better, but by 27%, instead of an order of magnitude.
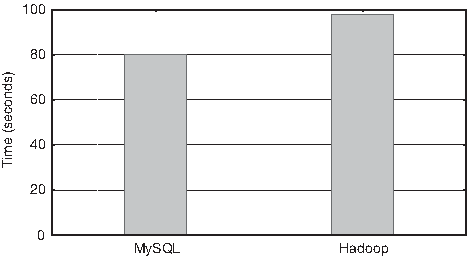
MySQL and Hadoop execution times when counting the total number of proteins for each organism.
Further similar experiments requiring combined accesses to various tables (i.e., Organism, Genestable, and Doctable) were performed and, in every case, MySQL performed better than Hadoop. These results indicate that a relational database like MySQL as the technology for implementing the database server for the current dataset and data-mining functionality of the Biblio- and Homol-MetReS applications is adequate, with their relatively simple and computationally inexpensive queries to the database.
3.3.2. Computing-intensive queries
In this section, we are interested in analyzing the performance of both data-handling frameworks when executing more complex analytical operations. The goal of this experiment was to calculate the frequency of the 20 different amino acids in the sequence of each protein of every organism. This permits an analysis of how protein and proteome complexity correlates to amino acid usage and allows users to investigate whether there are amino acids that are more frequently used in more complex proteins. To appreciate the performance difference with the previous experiment, Dataset2 was also used. The difference was mainly in the computing requirements, which are larger for the present experiment.
Algorithm 2 describes the main functionality of the test in the MySQL system, where a join query is performed to obtain the frequency of each amino acid. Equivalent Map (Algorithm 3) and Reduce (Algorithm 4) jobs were implemented for the Hadoop system. The map algorithm obtains the sequence field of each organism within the data, and the reduce algorithm counts each amino acid for each protein.
Figure 7 shows the execution times of both systems for this test. The Hadoop framework is 40% faster than the MySQL framework in this case. Additional similar tests were performed and led to similar results. Figure 8 shows the scalability of the Hadoop cluster when using from 8 up to 22 nodes. It can be seen how execution time significantly drops when increasing the amount of nodes. Taken together, these results show that Hadoop becomes a better alternative when the amount of data to be processed drastically increases and when the processing complexity grows.

Execution time with MySQL and Hadoop.
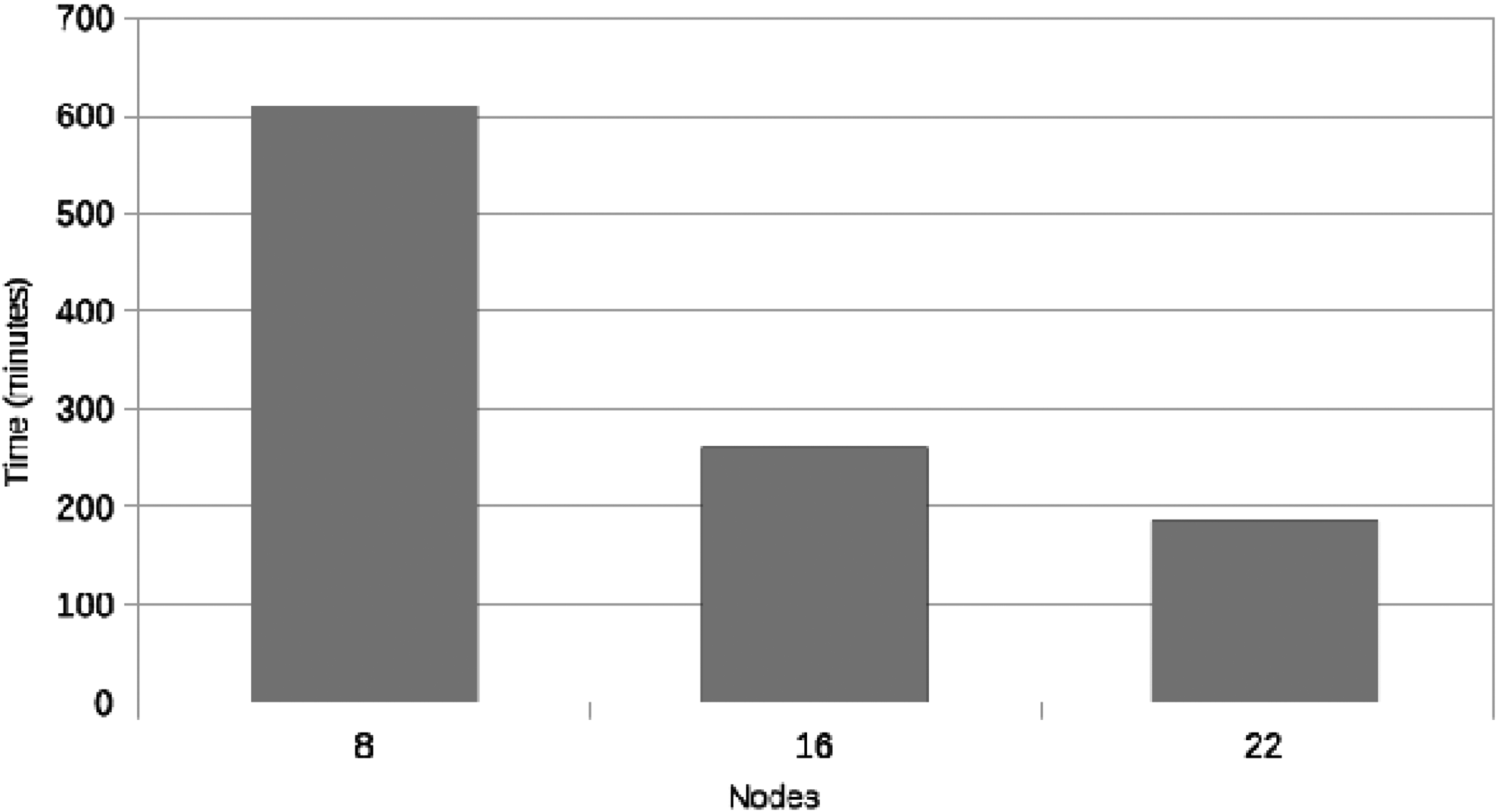
Execution time on the Hadoop cluster using 8, 16, and 22 nodes.
4. Discussion
These results illustrate the capacity of extracting valuable statistical analysis from our current dataset by using Big Data systems such as Hadoop, complementing the relational database used to support the web application. For small datasets and simple queries, the current optimized MySQL server offers better performance and is, therefore, most suited. However, when scaling the datasets by adding more organisms to the system and performing more complex analytic operations, the results show a significant performance improvement when using the Hadoop Big Data system. In this scenario, a 34.27% performance improvement over the previous MySQL system was obtained.
It has been proved that the Hadoop approach is not suited for replacing the MySQL system in all scenarios. It is a suitable complementary system that allows us to extract valuable information from our datasets more efficiently as they grow beyond what MySQL can appropriately manage and as the queries become more complex. Therefore, a hybrid approach where the MySQL server handles the most basic queries and the Hadoop server handles the most data-intensive ones is the best option for our specific system.
5. Conclusions and Future Work
The performance of two biological cloud-based applications was analyzed and optimized. A main alternative to the current database used by both applications was presented to obtain a better level of performance when scaling the data. The current database requirements were successfully analyzed, and the scenarios that could provide a greater positive impact on the system performance were determined.
The performed tests determined the characterization of queries and the dataset size in which a Hadoop-based deployment of the database in a cloud system may be needed. The results are satisfying and open a new range of possibilities for a deeper data analysis in both web applications, Biblio- and Homol-MetReS. As future work, we plan to implement such an analysis in the current cloud-based applications. Also, we expect to expand the database in the near future, which will allow us to perform further scalability experimentation with larger data sets.
Footnotes
Acknowledgment
This work was supported by the Ministerio de Economia y Competitividad under contract TIN2014-53234-C2-2-R and by the European Union FEDER (CAPAP-H6 network TIN2016-81840-REDT). The authors are members of the research group 2014-SGR163, funded by the Generalitat de Catalunya.
Author Disclosure Statement
No competing financial interests exist.