
Abstract
Abstract
The ability to efficiently sample structurally diverse protein conformations allows one to gain a high-level view of a protein's energy landscape. Algorithms from robot motion planning have been used for conformational sampling, and several of these algorithms promote diversity by keeping track of “coverage” in conformational space based on the local sampling density. However, large proteins present special challenges. In particular, larger systems require running many concurrent instances of these algorithms, but these algorithms can quickly become memory intensive because they typically keep previously sampled conformations in memory to maintain coverage estimates. In addition, robotics-inspired algorithms depend on defining useful perturbation strategies for exploring the conformational space, which is a difficult task for large proteins because such systems are typically more constrained and exhibit complex motions. In this article, we introduce two methodologies for maintaining and enhancing diversity in robotics-inspired conformational sampling. The first method addresses algorithms based on coverage estimates and leverages the use of a low-dimensional projection to define a global coverage grid that maintains coverage across concurrent runs of sampling. The second method is an automatic definition of a perturbation strategy through readily available flexibility information derived from B-factors, secondary structure, and rigidity analysis. Our results show a significant increase in the diversity of the conformations sampled for proteins consisting of up to 500 residues when applied to a specific robotics-inspired algorithm for conformational sampling. The methodologies presented in this article may be vital components for the scalability of robotics-inspired approaches.
1. Introduction
T
Algorithms from robot motion planning (Al-Bluwi et al., 2012; Gipson et al., 2012) constitute one such class of methods for enhanced exploration of the conformational space. For a great introduction to these kinds of methods, we refer the interested reader to Shehu and Plaku (2016). Robotics-inspired methods have been used for conformational sampling in a variety of cases, including protein folding (Amato et al., 2003; Thomas et al., 2007; Tapia et al., 2010), loop sampling (Yao et al., 2008; Shehu and Kavraki, 2012; Stein and Kortemme, 2013), identifying low-energy transitions between known conformations (Raveh et al., 2009; Haspel et al., 2010; Al-Bluwi et al., 2013; Gipson et al., 2013), exploring conformational space (Shehu and Olson, 2010; Jaillet et al., 2011; Gipson et al., 2013; Luo and Haspel, 2013), and improving fit to experimental data (Devaurs et al., 2016, 2017). These methods are characterized by their geometric reasoning to bias the exploration to unexplored regions of the space. Thus, the focus of these methods is on generating a diverse set of conformations.
Robotics-inspired sampling algorithms can be characterized by how they make two critical decisions. First, each algorithm must make a decision on where to sample in conformational space. These algorithms often estimate coverage based on the local sampling density to bias exploration toward less densely sampled regions of the conformational space. This is mainly how several robotics-inspired approaches promote structural diversity in their exploration. Second, once a selected region has been chosen, each algorithm must make a decision on how to generate a new conformation. Usually the proposed conformation is generated by perturbing a previously sampled conformation from the selected region and determining whether the proposed conformation is valid (typically by checking the energy or for steric clashes). In this work, we will use the term perturbation strategy to refer to how the algorithm generates new conformations from previously sampled ones. So, starting from an initial conformation, a robotics-inspired algorithm iterates over deciding where to sample, often using coverage estimates (first decision), then generating a new conformation based on its perturbation strategy (second decision).
Robotics-inspired approaches have seen many initial successes, whereas larger proteins present special challenges that complicate how these algorithms make the two critical decisions and, hence, hinder their ability to efficiently generate diverse conformations. First, larger proteins have more residues, or degrees of freedom, so sampling for such systems takes more time and memory and often requires running these algorithms concurrently across multiple cores. However, running many concurrent instances of robotics-inspired algorithms becomes memory intensive because of their frequent storage and use of previously sampled conformations as described by the first decision. In other words, robotics-inspired methods must be run for a shorter time as the size of the protein increases. As more concurrent instances are used, the memory usage rate increases since each instance needs to access all of the previous samples. The naive solution to this memory issue is to write the conformations to disk and restart conformational sampling at a randomly selected conformation, but we will demonstrate that this compromises the coverage estimates to the point where these algorithms lose the ability to promote diversity. So, in addition to the memory issue, there is also the problem of coordination across the concurrent runs of sampling.
Second, defining useful perturbation strategies becomes complicated for larger proteins because such systems are typically more constrained and exhibit complex motions. Larger proteins usually have high correlation among distant residues that results in more intricate movements. Perturbation strategies are then less likely to capture these movements, and the probability of proposing a valid conformation diminishes greatly (Vitalis and Pappu, 2009). Therefore, the ability to generate conformations that are highly different from the starting or reference conformation becomes challenging.
This has led to algorithms that aim at somehow encoding flexibility information into the perturbations (Shehu and Plaku, 2016). NMA-RRT (normal mode analysis with rapidly-exploring random tree) samples conformations by using normal mode analysis (Al-Bluwi et al., 2013). KGS (kino-geometric conformational sampling) uses sampling in the nullspace of the Jacobian of constraints to generate conformations that satisfy constraints introduced by hydrogen bonds (Pachov and van den Bedem, 2015). PCA-EA (principal component analysis with evolutionary algorithms) uses perturbations in a principal component space defined from wild-type and mutant structures, which are then translated to the original conformational space by using a combination of reconstruction algorithms (Clausen and Shehu, 2015). Note that defining perturbation strategies is also a problem outside of robotics-inspired methods as PCA-EA is an evolutionary algorithm. Our approach instead makes use of informed moves, which may be less computationally expensive because it does not require the use of reconstruction algorithms, computing Hessians (for NMA-RRT), or computing Jacobians.
In this article, we introduce two methodologies for maintaining and enhancing diversity in robotics-inspired conformational sampling. The first method addresses algorithms based on coverage estimates and leverages the use of a low-dimensional projection to define a global coverage grid that maintains coverage across concurrent runs of sampling. This global coverage grid keeps statistics about previously generated samples across different runs, which means that each run no longer needs to access all of the conformations. This approach solves not only the memory issue associated with robotics-inspired sampling methods that rely on coverage estimates but also the coordination problem of sampling across multiple cores. Our approach allows robotics-inspired methods to maintain the ability to efficiently decide where to sample conformations.
The second method is an automatic definition of a perturbation strategy derived from B-factors, secondary structure, and rigidity analysis. We also use the B-factor information to define the low-dimensional projection and compare with prior work on defining projections (Novinskaya et al., 2017). This method enhances diversity by focusing on how our algorithm perturbs conformations by using readily available flexibility information. Our results show that our methodology leads to a significant increase in the diversity of the conformations generated as well as the number of conformations generated for proteins consisting of up to 500 residues when applied to a specific robotics-inspired algorithm for conformational sampling, the Structured Intuitive Move Selector (SIMS) framework for conformational sampling (Gipson et al., 2013).
The rest of the article is organized as follows. In the next section, we describe our methodologies in detail, which are implemented into SIMS. In Section 3, we show how our new methodologies result in a significant improvement in the structural diversity of the sampled conformations. We also provide a discussion of the relative importance of each methodology. Finally, we conclude with a brief summary and a discussion of directions for future work.
2. Methods
2.1. Structured intuitive move selector
We apply our methods within the SIMS framework for conformational sampling (Gipson et al., 2013), which exemplifies the operation of several robotics-inspired methods through the combined use of the Open Motion Planning Library (OMPL) (Şucan et al., 2012) along with Rosetta (Leaver-Fay et al., 2011). In this section, we provide a high-level overview of SIMS that will introduce the components needed to describe our new methodologies. A detailed description of SIMS can be found in Gipson et al. (2013).
SIMS makes use of OMPL to determine where to conduct direct exploration through the use of coverage estimates, which are encoded inside a data structure called the coverage grid. The coverage grid data structure relies on a projection as input, which is used to map conformations to the coverage grid. The coverage grid consists of cells from the discretization of the mapped space, and each cell contains pointers to the conformations that are mapped to it. Deciding where to conduct direct exploration at any given iteration is a two-step process where (1) a cell is chosen and (2) a conformation is chosen within the cell. The probability that a cell is chosen is proportional to its computed importance value, which is a function of various grid cell statistics such as the number of conformations mapped to it. Essentially, cells that are less densely populated are chosen more often than cells that are more densely populated. Finally, a conformation is randomly chosen from the cell. More details are provided on the coverage grid in Section 2.2 and on the projection in Section 2.3.
SIMS determines how new conformations are generated through a so-called schema. The schema encodes how to repeatedly apply small perturbations called moves to previously generated conformations. SIMS uses an internal coordinate representation, where only dihedral angles are manipulated (bond angles and bond lengths are kept constant). SIMS' perturbation strategy, defined in the schema, specifies what type of moves to use, how they are applied, and how often to apply them. The moves are applied to sets of residues called fragments, where each move-fragment pair is assigned a weight to reflect how often to apply the move-fragment pair. The probability that a particular move-fragment pair is chosen at any given iteration is proportional to the weight. Ideally, the schema captures which fragments of residues might be involved in coordinated motion and how flexible they are (through the use of the weights). Previous work showed that secondary structure can be used to partition the protein into flexible loops, which should be perturbed more often (given higher weight) than relatively rigid helices and sheets (given lower weight) (Gipson et al., 2013).
For each type of fragment, different moves can be defined (e.g., loop sampling, random perturbation, energy minimization). SIMS makes use of Rosetta for implementations of the moves (Leaver-Fay et al., 2011). Once the move is applied to a fragment, side chain positions are determined by Rosetta's side chain minimization protocol (Das and Baker, 2008). Since SIMS uses Rosetta for the move implementations, SIMS' perturbation strategy can be easily extended to include advances in Rosetta's own robotics-inspired approaches to sample new conformations such as in Stein and Kortemme (2013). More details are provided in Section 2.3 on how a perturbation strategy in SIMS is constructed.
Each proposed conformation is automatically rejected if the computed energy of the proposed conformation is above a user-defined threshold. In this work, we use Rosetta's “score12 full” all-atom energy function for our smaller-sized proteins and Rosetta's “score3” energy function in “centroid” mode for our larger-sized proteins, although other energy functions could be used as well. Centroid mode computations in Rosetta are faster because side chains are approximated as a single atom of varying size, which provides additional computational benefit for larger proteins while still maintaining molecular detail. Energy thresholds are chosen to filter out conformations with steric clashes and other highly unfavorable interactions. Energy thresholds for this work are set to the value 0 because past experiments tend to show that conformations with a positive Rosetta score have some degree of steric clashes. One could always lower the energy threshold or filter out high-energy conformations in a post-processing step to obtain sampled conformations with lower energy.
2.2. Global coverage grid
The first critical decision that robotics-inspired methods have to make is where to direct the exploration. Many robotics-inspired methods, such as SIMS, base this decision on the computed coverage estimates. Coverage estimates measure where the less-densely sampled regions of the conformational space are located. Based on the coverage estimates, robotics-inspired approaches incorporate a bias toward the unexplored regions of the space (Shehu and Plaku, 2016). Computing coverage estimates in robotics-inspired methods becomes complicated for larger systems, so in this section we describe a method that can maintain their ability to compute coverage.
Conformational sampling generally becomes a harder problem as the size of the considered system increases. Unless the system is highly stable and only exhibits small-scale movements (like side-chain rearrangements), more computational resources are needed. Energy computation takes longer so we must run simulations longer to sample a given number of conformations. The conformational space is also larger so we may need more conformations to accurately characterize the space. These complications give rise to the need to run multiple robotics-inspired sampling across many cores. However, keeping all of the sampled conformations in memory means that the rate of memory usage increases. For the rest of this article, we will refer to this as the “memory issue.” Although there has been work on parallelizing robotics-inspired sampling techniques (Plaku et al., 2005; Ichnowski and Alterovitz, 2012; Devaurs et al., 2013), these approaches do not address the fact that memory use becomes a bottleneck for large proteins. So in this work, we are addressing the problem of running multiple instances of SIMS concurrently in an efficient manner that can also handle the memory issue.
Initially, one may consider keeping all of the sampled conformations on disk and running separate, concurrent instances of SIMS across each computing core. But this means that each core must have access to all of the sampled conformations because any previously sampled conformation could be perturbed in a given iteration. This may be expensive if every iteration involves reading and writing to disk. We could also write all the conformations to disk periodically and restart the exploration from randomly chosen points. However, as mentioned in Section 1, this results in losing the vital coverage estimates. After a restart, the algorithm is likely to re-explore parts of conformational space that have been densely sampled in previous runs.
Keeping all of the sampled conformations on disk appears to be unavoidable when running multiple instances of SIMS for a long time. So to prevent excessive reading and writing to disk, each instance of SIMS must work with its locally generated set of conformations. The question becomes how each instance of SIMS can “get informed” of the work that other instances of SIMS are performing. We solve this by leveraging SIMS' use of a low-dimensional projection to keep coverage estimates and implementing a global coverage grid, whose scope reaches across all the instances of SIMS.
In SIMS, sampled conformations are added to the coverage grid data structure through the use of a projection (detailed in Section 2.3). The grid contains cells with conformations mapped to them and by counting how many conformations map into each cell, we can estimate the sampling density or coverage. Different robotics-inspired techniques such as Expansive Space Trees (Hsu et al., 1999) and Kinodynamic Motion Planning by Interior-Exterior Cell Exploration (KPIECE) (Sucan and Kavraki, 2009) use this information to guide the sampling toward less-densely sampled parts of the conformational space. In this work, we use KPIECE since it has been shown to significantly outperform EST (expansive-spaces tree planner) (Sucan and Kavraki, 2009).
KPIECE keeps track of various statistics for each grid cell and uses these statistics to compute a heuristic called importance for each cell. Conformations in cells with higher importance are perturbed more often. Importance is computed for each cell by using four statistics:
(1) The number of projected conformations mapped into the cell. (2) The number of times the cell has been chosen for expansion. (3) The iteration in which the cell had its first conformation mapped to it. (4) The number of cell neighbors that have conformations mapped to it.
An increase in (1), (2), and (4) produce lower importance whereas a high value for (3) produces greater importance. Indeed, these are the statistics that are lost when conformations must be written to disk. Fewer conformations are accounted for in the coverage grid, and the importance heuristic loses the ability to differentiate cells based on sampling density.
Our method saves global grid cell statistics into a central database along with conformations sampled from each SIMS instance. In this work, we used an MySQL database to handle the read/write requests from the multiple SIMS instances, and we created tables to simply hold the conformations and global coverage grid. When a SIMS instance is started, a subset of conformations is loaded along with “summarized” coverage statistics about the global coverage grid. These “summarized” coverage statistics are an indirect way of accounting for the sampling done by other SIMS instances. The SIMS instance then proceeds for a specified amount of time. The SIMS instance maintains its own local coverage grid (using the conformations generated by the run) enriched with the summarized coverage statistics (taken from the global coverage grid). When an SIMS instance is ready to write conformations, the new conformations are written to the database, and the global grid cell statistics are then updated. Thus, our method provides the coordination across the SIMS instances that effectively maintains coverage estimates.
Grid cell statistics on the global coverage grid are maintained centrally in a similar manner to how an individual SIMS instance computes grid cell statistics on a local coverage grid. Each grid cell computes an importance heuristic that determines how often conformations from that cell are perturbed. In the context of the global coverage grid, (3) is no longer used to compute importance because there is no meaningful way to define iteration when multiple cores are sampling simultaneously. However, (1), (2), and (4) are still used.
When an SIMS instance is finished, the new conformations are written to the database and the global grid cell statistics are updated. This is done by computing the change in (1) and (2) during the course of the SIMS run. These values are then added to the global values of (1) and (2) in the database. (4) is subsequently updated based on the new grid cell statistics. These statistics are global in the sense that all the instances provide an update when their run is finished.
When an SIMS instance is restarted, the KPIECE sampling strategy is used to select new starting conformations based on the global coverage grid. In addition, the local coverage grid is initialized to the current values in the global coverage grid. Although each core is aware of the global coverage statistics, each core can only perturb conformations that are in memory (i.e., generated since the start of the SIMS instance). However, when a conformation is generated in a cell, the sampling density is determined not only by the conformations generated from the SIMS instance but also by the sampling density loaded at the start of the run (Fig. 1). Thus, the presence of all other sampled conformations is accounted for indirectly.

Pictorial representation of the effect that the use of summarized coverage statistics has on a local coverage grid. Each blue dot represents a previously sampled conformation. Before hitting memory limits, the exploration is proceeding in some direction depicted by the black arrow. When the exploration restarts, summary coverage statistics are maintained and depicted in shades of gray. Darker shades indicate more densely sampled areas that the algorithm can use to reduce sampling redundancy.
We now claim that this algorithm also solves the memory issue. Each core will cycle through three steps: reading conformations and statistics from the database, running an SIMS instance, and finally writing conformations and updating the global statistics. The frequency in which each core does this process is a user-defined parameter called the restart frequency. The memory issue is avoided through the use of the summarized coverage and a restart frequency that is not too low. That is, we must restart often enough such that a core will not run out of memory from sampling too many conformations. Interestingly, there is incentive to restart often as this is the mechanism in which the database is updated with the work that each SIMS instance has done. On the other hand, saving conformations to a central database and restarting has some computational overhead associated with it. Our experiments use a restart frequency of 6 per hour (restart every 10 minutes). We leave it as future work to determine an optimal value for this parameter.
Finally, we note that when an instance syncs with the database, then the information in the coverage grid could not be as up to date as possible. There could be other instances that have explored parts of conformational space that the coverage grid is not yet aware of. Our results in Section 3 indicate that this is not a cause for concern. Simply having some notion of the work done by other instances is enough to see an improvement in the diversity of conformations generated using our methodologies. The reason for this is because the original KPIECE method (Sucan and Kavraki, 2009) is inherently adaptive. If at some point of execution multiple cores are working in the same region of the conformational space, the global coverage grid will eventually get “informed” of the work when the cores synchronize with the database. Then when new SIMS instances are created, these instances are less likely by design to work in the same region again because of the importance values in the coverage grid.
2.3. Defining a perturbation strategy using flexibility information
The second critical decision that robotics-inspired methods have to make is how to generate new conformations. In other words, these methods have to define a perturbation strategy to obtain new conformations from previously sampled ones. Defining perturbation strategies for larger proteins is more difficult because of the high correlation among residues, and naive/uninformed strategies will result in high rejection rates. In this section, we will describe how to generate informed perturbation strategies through readily available flexibility information. In the context of SIMS, this translates to finding a definition of the schema. Note that although we focus the construction of the new perturbation strategy on the schema used in SIMS, the same ideas can also be incorporated into other conformational sampling frameworks. We show how to automatically generate a schema that biases perturbations toward fragments that are more flexible by using a combination of B-factors, secondary structure, and rigidity analysis.
The new perturbation strategy incorporates global structure information into the schema. In previous iterations of SIMS, the default schema made use of only secondary structure information. Alpha helices and beta sheets were made more stable than loops. However, secondary structure is essentially local information because every secondary structure element consists of a few consecutive residues. In general, helices and sheets are more stable than loops, whereas helices and sheets from different parts of the protein may have vastly different flexibility (Novinskaya et al., 2017). The old default schema essentially lacked tertiary structure information describing global flexibility.
Secondary structure is readily computed or available in the PDB, whereas tertiary structure information is not available directly from experiments. However, an approximation can be derived from the atom coordinates using rigidity analysis. This is done with KINARI web, a suite of tools for computing rigidity and flexibility of biomolecules (Fox et al., 2011). KINARI web uses the pebble game algorithm to compute clusters of residues that are expected to move together (Lee and Streinu, 2008). A PDB file is inputted into the web server to get the residue clusters. All default parameters are used in the computation. KINARI outputs a file that specifies residue clusters. Each cluster consists of a set of residue intervals. We use each residue interval from each cluster as a separate residue grouping. These residue groupings will be used by the schema to model parts of the protein that are supposed to move together. We could have chosen to instead use the cluster of residues (or a set of intervals) as the residue groupings for a simpler rigidity model, but KINARI may only detect one or two residue clusters. Our definition of the residue groupings allows for a more fine-grained decomposition of the protein.
The schema used in SIMS specifies fragments, which consist of groups of residues, and moves, which define perturbations on the fragments. Each fragment is assigned a weight that describes how frequently the fragment is chosen to be perturbed. SIMS currently has five major moves that are briefly defined as follows:
(1) Minimization involves a few steps of an energy minimization protocol on the fragment. We use the “dfpmin_armijo_nonmonotone” protocol (www.rosettacommons.org/docs/latest/rosetta_basics/structural_concepts/minimization-overview) and run until a tolerance of 0.01. (2) Loop sampling involves sampling a random loop conformation with the constraint that the endpoints remain in the same position. (3) Rigid-body sampling involves rotating and translating one part of a domain relative to another. This is done by a displacement of one loop endpoint relative to the other. (4) Random single perturbation involves randomly perturbing a single residue's dihedral angles within a given fragment. (5) Randomize all involves perturbing all the dihedral angles in a given fragment.
In addition to the rigidity analysis, which has been used earlier in robotics-inspired sampling (Thomas et al., 2007; Luo and Haspel, 2013; Andersson et al., 2016), our method assigns a weight to each fragment by using B-factors. SIMS relies on a starting conformation that is derived from experiments. These experiments will have some measure of uncertainty, which is usually correlated with flexibility or movement. For X-ray crystallography experiments, B-factors (also known as temperature factors) describe the displacement of the atomic positions from their mean values (Trueblood et al., 1996). These B-factors can be easily extracted from a PDB file (coordinates of a protein conformation derived from experiment) to generate the projection. B-factors can also be generated from prediction tools (Yuan et al., 2005). As a reminder, the probability that a move-fragment pair is chosen is proportional to its assigned weight. Thus, using B-factors as the weights naturally biases the fragments that are more flexible.
For a system with n residues, n B-factors corresponding to the alpha carbon atoms in the backbone are extracted from the PDB file. Then for each factor bi,
This transformation is done because we are only interested in the relative flexibility of the fragments to each other. B-factor data may contain noisy values for fragments that may overly dominate and cause the method to over sample this region. For our experiments, we usually set
This transformation essentially spreads the values farther apart from one another. The amount of spreading can be controlled by using another user-defined parameter
Using KINARI, secondary structure information, and B-factors, the schema can be automatically generated. We propose to generate a schema that consists of three major classes of fragments. The first is a class containing only a single fragment that is defined over the whole protein. This class is sampled 10% of the time and is used to occasionally generate structures with a lower energy (minimization) or try disruptive whole protein perturbations (rigid-body sampling). When this fragment is sampled, minimization is chosen 90% of the time and rigid-body sampling is chosen 10% of the time.
The second major class of fragments is generated by using secondary structures. This class of fragments constituted the majority of the fragments in previous iterations of SIMS. We place less overall influence on this class since we only sample this set 40% of the time (compared with 90% previously). The secondary structure information is extracted from the PDB file. Alpha helices and beta sheets are treated as loops if they are less than 5 residues in length. A fragment is then defined for each consecutive interval of residues with the same secondary structure classification. Loops can be perturbed by using loop sampling (10%), random single perturbation (30%), randomize all (30%), or rigid-body sampling (30%). Helices and sheets are generally more stable, so we use random single perturbation (50%) or rigid-body sampling (50%). In previous work, SIMS manually defined loops to be sampled with greater weight than helices and sheets. Fragments are sampled with a weight computed from the B-factors described earlier.
Finally, the third major class of fragments is generated by using the residue groupings from KINARI. Since each residue grouping is predicted to move together, a fragment is defined for each interval of residues between the residue groupings (intervals at the ends are not counted). In other words, the parts of the protein that we wish to perturb are the residues that lie in between tertiary structures, which we call hinges in this work. Note that this work uses hinges in a different manner as is used in the rigidity analysis community. When these hinges are perturbed, the surrounding parts move together as a rigid body. Each fragment is weighted by using B-factors and can be perturbed by using loop sampling (10%), random single perturbation (35%), randomize all (20%), or rigid-body sampling (30%). This class of fragments is sampled 50% of the time.
The increased emphasis on tertiary structures is more aligned with the intended use of SIMS for sampling large backbone motions. All of the percentages given earlier were determined empirically, and further research may fine tune these values.
Finally, recall that SIMS uses a projection that maps a conformation to the coverage grid. The projection can be automatically generated by using B-factors. For a system with n residues, the projection will be of dimension
The other
3. Results
Our main objective was to determine the effect our new methodologies had on the diversity of the conformations sampled. All of our experiments are run on a single compute node with two Intel E5-2650v2 Ivy Bridge EP processors for a total of 16 cores, where each core runs an instance of SIMS. All runs are done for 100 minutes and write conformations to disk every 10 minutes (restart frequency is 6 restarts per hour). Energy thresholds for all the experiments are set to the value 0 because past experiments tend to show that conformations with a positive Rosetta score have some degree of steric clashes. All the projections used are two-dimensional (2D). In the Discussion section, we call the version of SIMS with the new methods, “SIMS 2.0,” which includes the global coverage grid implementation along with the new perturbation strategy defined in the schema as described in the previous section. The version without the new methods is called “naive SIMS.”
3.1. Proteins used in experiments
We illustrate the benefits of our new methods on four proteins of varying sizes: cyanovirin-N (CVN) (Botos et al., 2002), calcium-loaded calmodulin (CaM) (Anthis et al., 2011), ribose-binding protein (RBP) (Björkman et al., 1994; Björkman and Mowbray, 1998), maltodextrin-binding protein (MBP) (Quiocho et al., 1997), and a single subunit of GroEL (Skjaerven et al., 2011, 2012). CVN, CaM, RBP, and MBP are smaller sized proteins that we have previously studied in the context of evaluating random projections (Novinskaya et al., 2017). The GroEL subunit is a larger and more constrained system consisting of about 500 residues. We also depict the B-factors and the KINARI information onto the structures to give a sense of how the new method is defining in the schema.
3.1.1. Cyanovirin-N
CVN is a 101-residue bacterial protein (PDB 3EZM) that exhibits antiviral activity toward the human immunodeficiency virus. CVN shows large-scale motions from the correlated activity of three loop regions at residues 24–28, 50–55, and 75–80. These same loop regions are found as flexible from Figure 2a. From Figure 2b, KINARI classifies two of these loop regions (residues 24–28 and 40–54) as hinges (which we defined in the previous section as the residues in between residue groupings). When constructing the schema as described in the previous section, the range of the B-factors is 10–20, instead of the default 20–50.
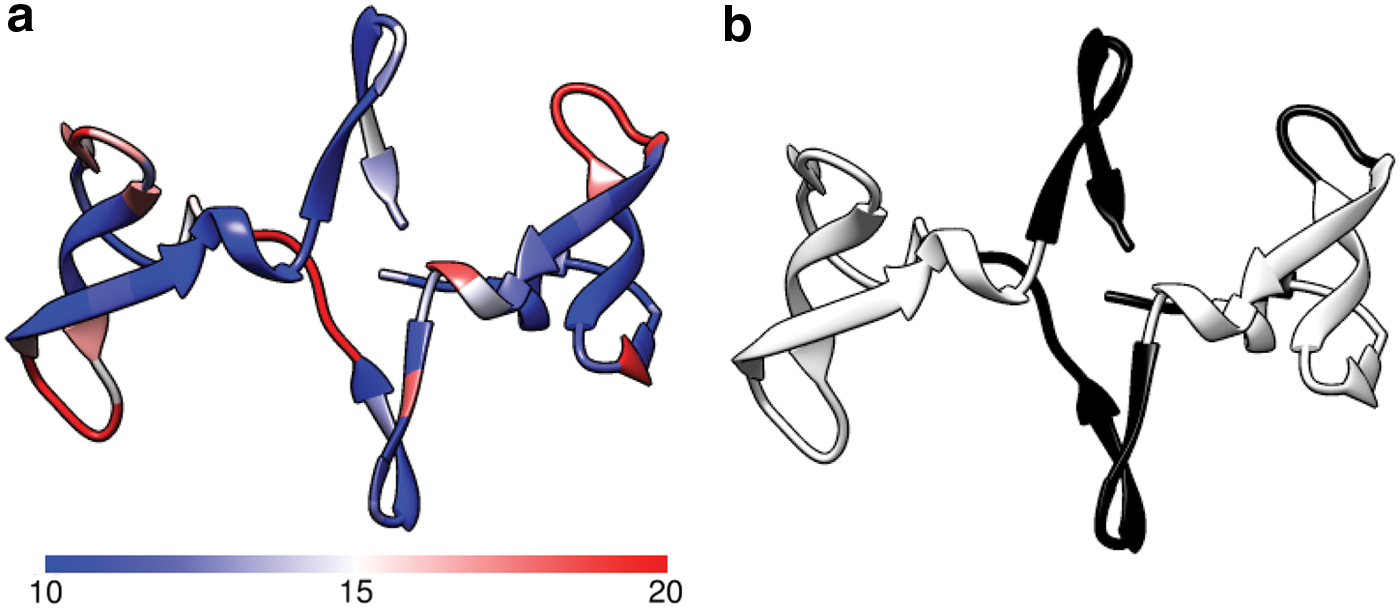
Cyanovirin-N.
3.1.2. Calmodulin
CaM is a 144-residue protein (PDB 1CLL) involved with interactions between calcium ions and other proteins. B-factors show that the flexible parts of the protein are found in residues 5–20, 35–41, 52–57, 67–80, 87–93, 107–116, and 126–129, which are represented in Figure 3a. Note that the flexible helix at residues 67–80 would have been treated as a more stable part of the structure (and hence, not perturbed frequently) if only secondary structure information was used. The computed hinges in Figure 3b are loops located at residues 41–43, 57–62, and 114–116.

Calmodulin.
3.1.3. Ribose-binding protein
RBP is a 271-residue protein (PDB 1URP, chain A) that consists of two domains connected by three loop regions located at 91–104, 226–237, and 253–269. The first two regions are more constrained and have to move in a coordinated way. Interestingly, the B-factor distribution in Figure 4a shows that the most flexible parts are mainly the alpha helices at the end. The KINARI output in Figure 4b also predicted that most of the protein move together (residues 3–205 and 211–268), leaving a single hinge at residues 206–210 that is not part of the three main loop regions. Nevertheless, the B-factors for the main loop regions, indeed, show greater flexibility than the surrounding two domains.
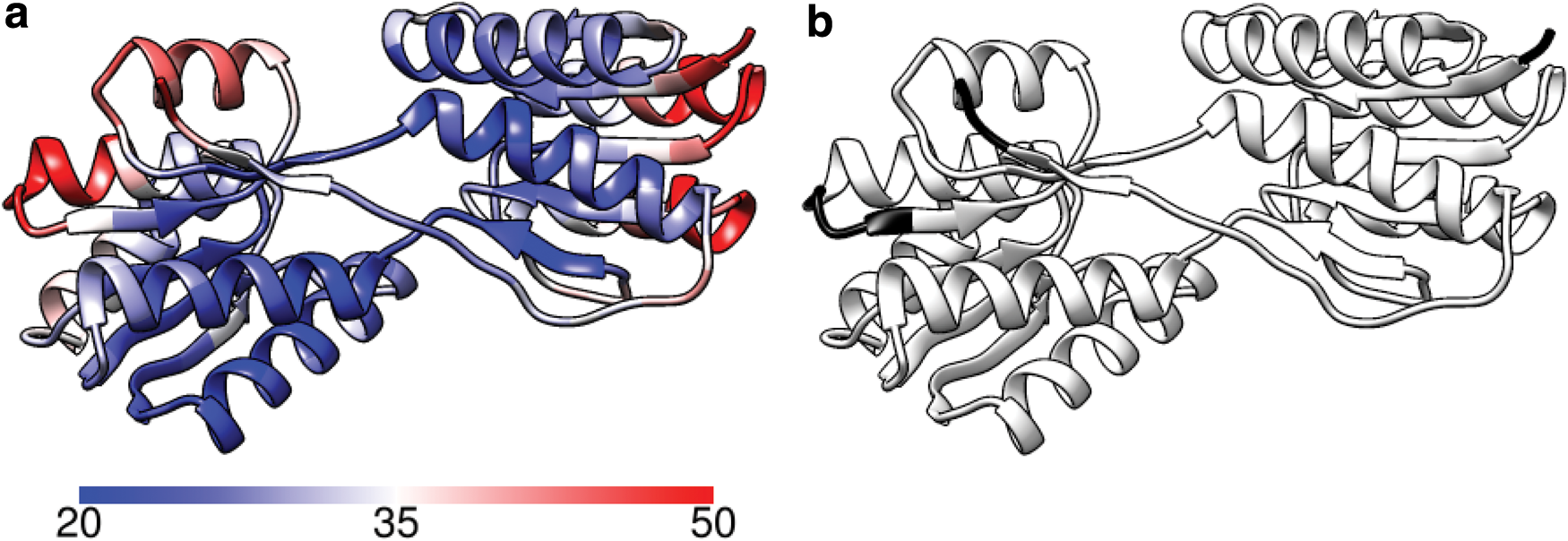
Ribose-binding protein.
3.1.4. Maltodextrin-binding protein
MBP is a 370-residue protein (PRB 3MBP) that consists of two domains on each end terminal. MBP is known to exhibit protein-wide conformational changes between the open and bound forms that involves movement in nearly all the residues. The B-factor distribution in Figure 5a shows most flexibility at the extreme ends of the protein, which will allow the schema to focus on the two domains. However, KINARI predicted that most of the protein is rigid, so there is only a single major hinge shown in Figure 5b.
Maltose-binding protein.
3.1.5. GroEL subunit
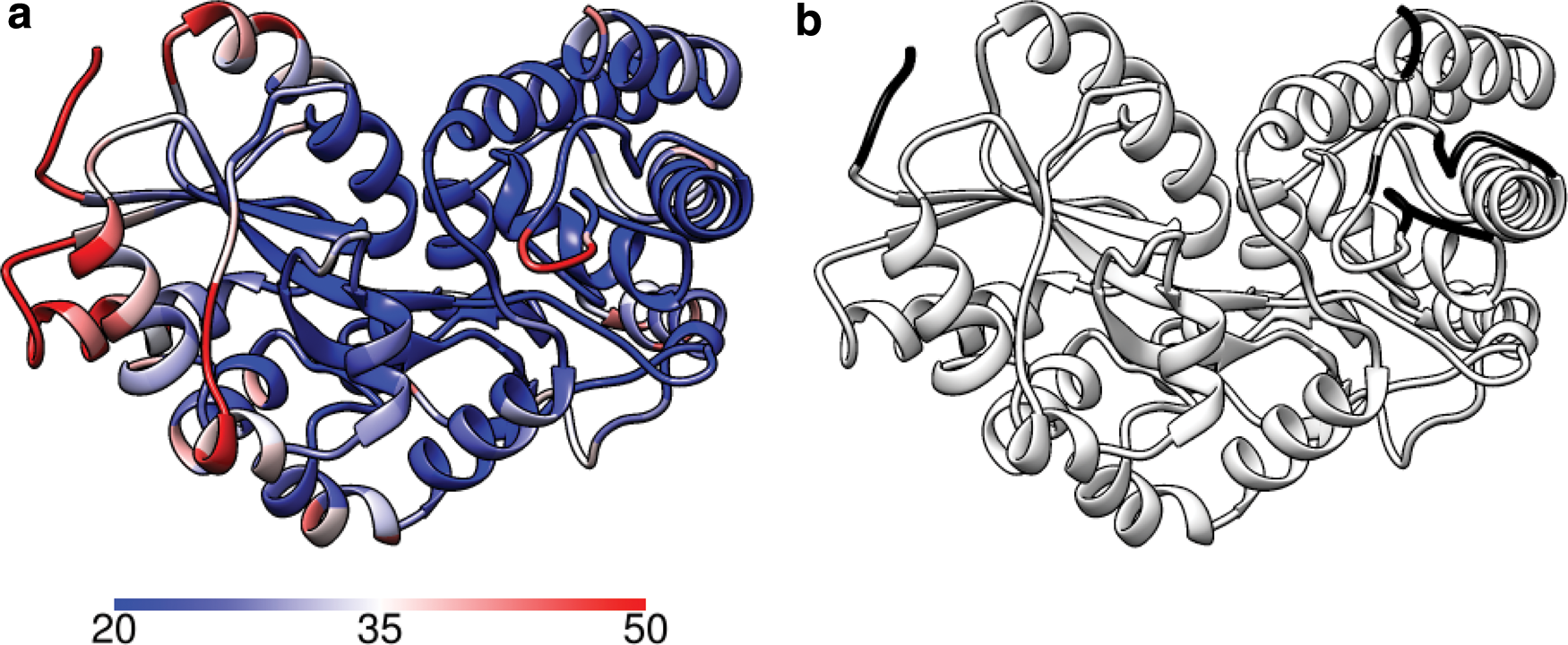
GroEL (PDB 1XCK) is a molecular chaperone consisting of 14 identical subunits forming two heptameric rings. We extract out chain A and use this as input to KINARI web. Each subunit consists of 524 residues arranged into three domains (Fig. 6a): equatorial (1–133, 409–524), intermediate (134–190, 377–408), and apical (191–376). The apical domain has the most movement, facilitated by hinges located in the intermediate domain (Skjaerven et al., 2011, 2012). The equatorial domain remains mostly stable.

A single subunit of GroEL.
In Figure 6b, notice that the high B-factors correlate to the apical domain, which is known to be the most flexible part. Figure 6c shows the parts of the subunit from which we treat as hinges. Note how the hinges are mostly loops located between major helices/sheets in the system. Perturbing these hinges will, in turn, affect the alpha helices and beta sheets, analogous to a rigid body transform.
3.2. Increased number of generated conformations
In this section, we want to get a sense of how many conformations SIMS 2.0 can produce given a certain number of cores. With conformational sampling for larger systems, the energy computation becomes more expensive, so the rate at which conformations are produced becomes vital. Producing more conformations is more efficient in the sense that the method is rejecting conformations less often. Note that all of the conformations produced are required to be below an energy threshold. We run experiments on a varied number of cores (1, 4, and 16) to assess the effect that our new methods have on scalability. Table 1 records the average number of conformations produced.
Standard deviations shown in parentheses. As the number of cores increase, the new version of SIMS produces conformations at a faster rate.
CaM, calmodulin; CVN, cyanovirin-N; MBP, maltodextrin-binding protein; SIMS, Structured Intuitive Move Selector; RBP, ribose-binding protein.
Table 1 clearly shows that as the number of cores used increases, the difference in the number of conformations produced by SIMS 2.0 compared with naive SIMS increases. Note that all of the conformations produced are below the user-defined energy threshold. For a given number of cores, the rate at which conformations are being produced by SIMS 2.0 is greater than naive SIMS. Hence, the rejection rate of the proposed conformations is lower in SIMS 2.0. As discussed earlier, high rejection rates in sampling for larger proteins was an issue preventing the scalability of robotics-inspired approaches (in how conformations are generated). SIMS 2.0 would make more efficient use of resources when the search requires up to 16 cores.
3.3. Improved conformational space coverage
The results from the previous section say nothing about the diversity of the conformations produced. In this set of experiments, we fix the number of cores to 16 and assess how well SIMS 2.0 generates diverse conformations. We use
3.3.1. Nearest neighbor distances
We first measure the closeness of the conformations from each other. This is done by tracking the distance of each conformation to its nearest neighbor. This is a measure of how “spread out” the conformations are from each other. If the conformations are all similar to their neighbor, then the algorithm may not have produced a structurally diverse set of conformations. Another way to interpret a small value using this measure is that neighboring conformations are more likely to have been sampled next to each other (one conformation was perturbed to get the other). So if the average nearest neighbor distance is higher for one method, then the average effect of each perturbation was greater and, hence, produced a more rapid exploration. From Figure 7, we see that SIMS 2.0, indeed, produces conformations that are farther apart from each other compared with naive SIMS.
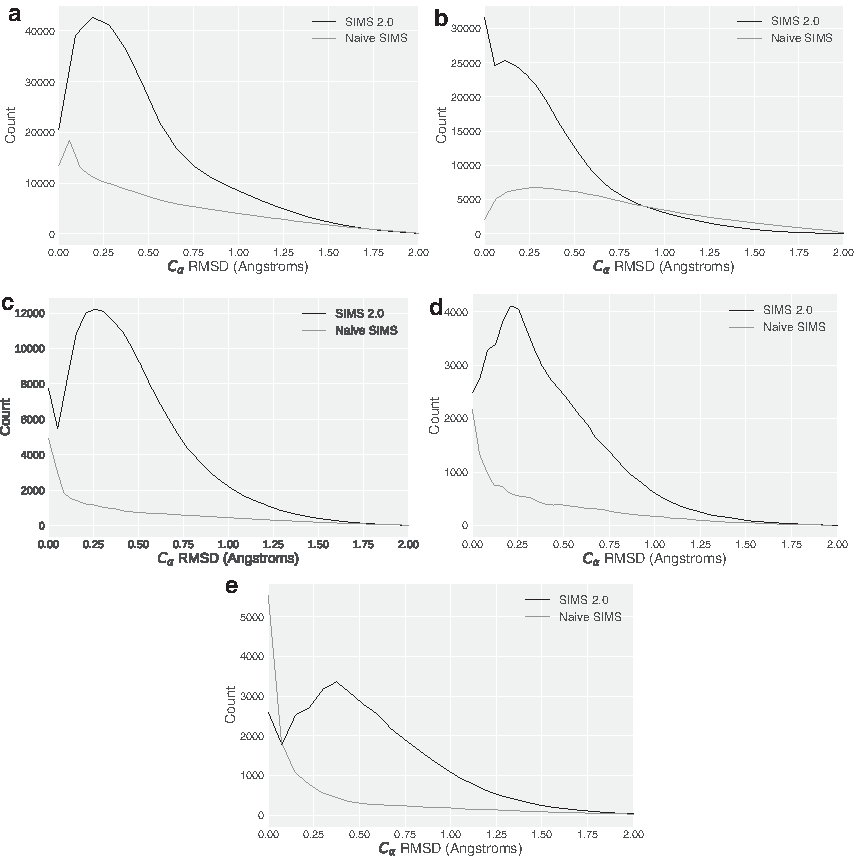
Density of nearest neighbor distances (averaged over 10 runs) for CVN
3.3.2. Expansiveness
We now measure the expansiveness of the conformational search. A more expansive search means that farther parts of the conformational space are sampled given the same starting point. Figure 8 shows a density plot of the distances of each conformation from the start conformation. The same start conformation was used for these experiments for a given protein. In addition to producing more conformations, SIMS 2.0 also produces more conformations that are farther from the start. Therefore, SIMS 2.0 produces a more expansive search than naive SIMS.
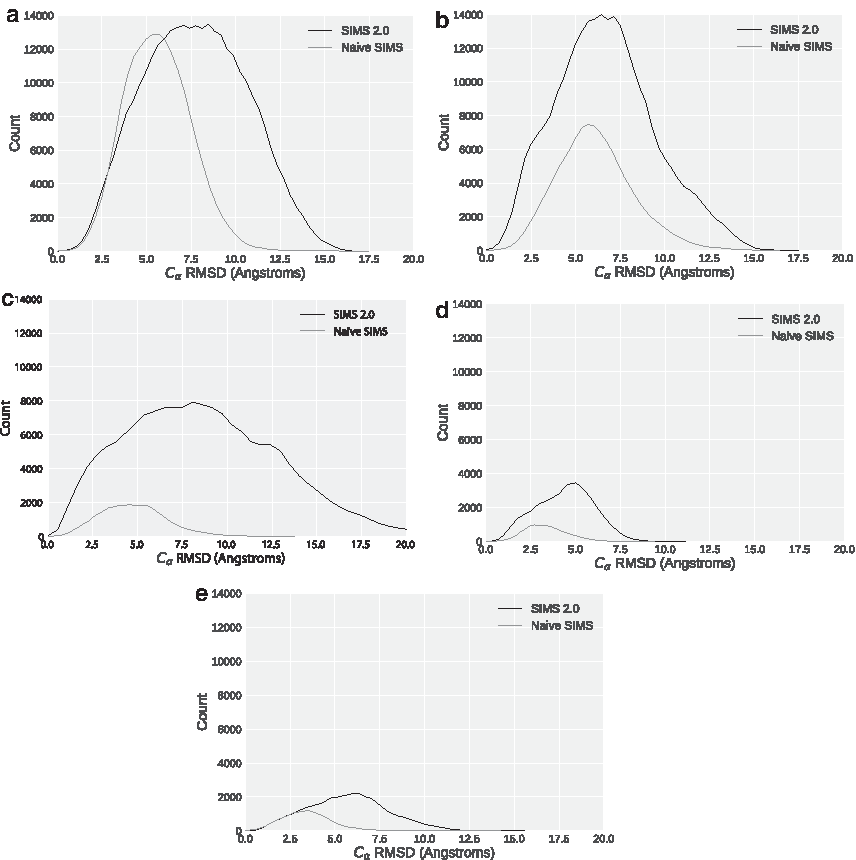
Density of distances to start conformation (averaged over 10 runs) for CVN
3.3.3. Isolating each improvement
These results taken together show that SIMS 2.0 produces more diverse conformations with the new methods compared with naive SIMS. We now end this section by investigating which specific method contributed most to the improved expansiveness. We focus the following experiments on the GroEL subunit system. Results for the other proteins were qualitatively similar.
We first ran an experiment with SIMS 2.0, where the global coverage grid did not send summarized coverage estimates when an SIMS run restarted. This run is similar to naive SIMS except with the new perturbation strategy. Figure 9 shows the effect this had on expansiveness. It appears that since no synchronization was occurring, the exploration was not as expansive, which was likely due to the increased amount of repeated work done among the cores.
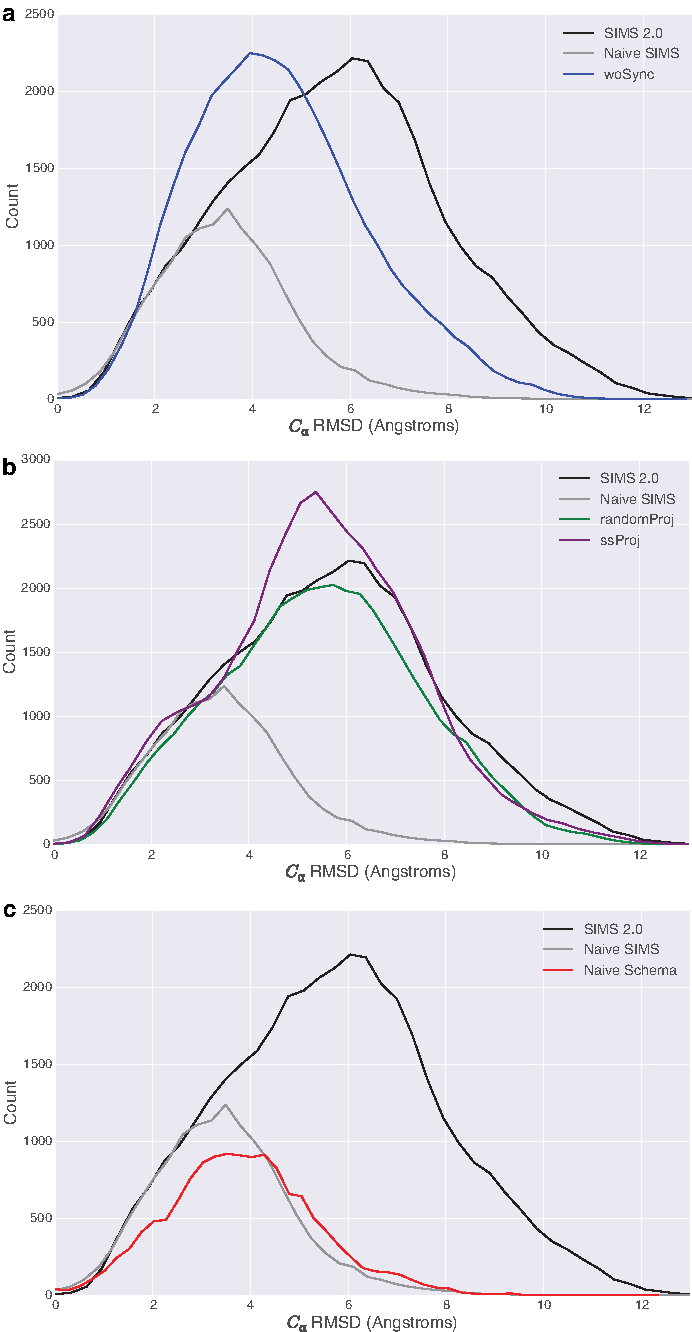
Density of distances to start conformation (average over 10 runs) for the GroEL subunit. (Top) woSync (blue) corresponds to the runs where the synchronization of coverage statistics was turned off. Since the distribution shifted to the left, the exploration was less expansive than SIMS 2.0. (Middle) randomProj (green) corresponds to the runs where a random projection was used. ssProj (purple) corresponds to the runs where secondary structure information was used to construct the projection. Since the distributions barely shifted, the definition of the projection does not appear to have much impact on the expansiveness. (Bottom) The new schema appears to have the greatest impact since the run that uses a schema with only secondary structure information (Naive Schema, red) has a density curve that is most similar to naive SIMS (old, gray). SIMS, Structured Intuitive Move Selector.
Next, we focus on the projection defined by B-factors. We ran two additional experiments. The first is SIMS 2.0 using a random projection (randomProj) (Gipson et al., 2013). The second is SIMS 2.0 using a projection constructed from secondary structure (ssProj) as mentioned in Novinskaya et al. (2017). The results in Figure 9 imply that the projection definition is not vital to the exploration. The exploration using a random projection only appears to be marginally worse than the one from SIMS 2.0. In addition, the projection using secondary structure information does not provide much improvement over the runs using a random projection. This is likely due to the fact that we are using a linear, 2D projection to represent a complex, high-dimensional conformational space. Thus, even though we use B-factors to incorporate flexibility into the projection definition, this translates to a relatively small improvement in how coverage is computed since the space is so simplified and much information is lost in the projection operation.
Finally, we focus on the schema improvement. We ran an experiment with SIMS 2.0 by using a schema defined using only secondary structure information. The new schema appears to contribute the greatest since the density curve with a naive schema is most similar to the one corresponding to naive SIMS even though the other improvements are included. The density curve with a naive schema also implies that the improvement in the number of conformations produced was also due to the new schema, since the height of the density curve of naive schema is lower than the one from SIMS 2.0. This demonstrates how important the schema is to the exploration because it essentially encodes how SIMS 2.0 explores the space.
4. Discussion
We have shown that with the addition of the global coverage grid and a perturbation strategy enriched with flexibility information, SIMS 2.0 can more efficiently generate conformations that are also distributed more diversely as compared with naive SIMS. All of the comparisons done were under the same computing budget, and the conformations generated were all under the same energy threshold. These new methodologies present promising steps toward making robotics-inspired conformational sampling better suited for larger proteins. Although the example proteins ranged from about 100 to 500 residues, additional work may be required for proteins that are greater than 500 residues in length. Since our results show that the construction of the perturbation strategy contributed most to the improvement in structural diversity, we may have to adjust the parameters in the schema construction to handle even larger systems. For example, the KINARI results for RBP and MBP did not provide any significant decompositions of the protein that the schema could exploit, and perhaps adjusting the parameters in the KINARI software could provide a better set of residue groupings. Our results also show that for a given number of cores, SIMS 2.0 produces conformations at a faster rate than naive SIMS. This points to the additional benefit of adding flexibility information to the perturbation strategy. Our experiments only went up to 16 cores to represent more modest computational resources such as a high-end desktop. However, more work is needed to characterize how SIMS 2.0 performs in a large-scale setting with hundreds of cores.
5. Conclusion
Robotics-inspired approaches rely heavily on two critical decisions: where to focus sampling in the conformational space and how to sample new conformations. As the protein size increases, robotics-inspired methods that run across multiple cores become memory intensive, coverage estimation becomes more expensive to maintain, and the definition of a useful perturbation strategy becomes difficult. In this article, we introduced two methodologies to maintain and enhance the diversity of conformations sampled and implemented these in SIMS. First, we proposed to maintain a global coverage grid that eliminates the memory bottleneck for large proteins and enables efficient conformational sampling across multiple cores. Next, we presented a perturbation strategy by using flexibility information from B-factors, secondary structure, and rigidity analysis.
For SIMS, our results show a significant improvement in the diversity of the conformations generated with our methods for proteins consisting of up to 500 residues. We also showed that our methods increased the number of conformations generated at a faster rate as the number of cores increased. We demonstrated that the new perturbation strategy provided the most dramatic change in diversity in terms of expansiveness (as measured in terms of distance from the start conformation). Our methods solve both the memory problem associated with SIMS and the coordination problem of sampling across multiple cores. Our new perturbation strategy is also a practically free way to obtain informed moves that improve structural diversity over a simple, naive strategy.
For future work, we plan to apply these ideas to other robotics-inspired conformational sampling frameworks. The ideas introduced in Section 2.2 apply to robotics-inspired methods that keep track of coverage, whereas the ideas in Section 2.3 apply more generally to robotics-inspired methods. Also, we will work on improving SIMS 2.0 through a variety of directions. We can combine our global coverage grid with other existing perturbation strategies. Further, we will also begin to investigate new ways to keep track of coverage. Our experiments showed that the currently used projection definition does not greatly affect the expansiveness of the exploration, so we have begun to look at non-linear projections. This work also showed that the most significant improvement in terms of diversity came from the new perturbation strategy. We will investigate the benefits of a dynamically changing perturbation strategy that perturbs conformations differently as a function of the currently chosen conformation.
Footnotes
Acknowledgments
This work is supported in part by NSF CCF 1423304, Rice University funds, and a training fellowship from the Gulf Coast Consortia on the Training Program in Biomedical Informatics, National Library of Medicine T15LM007093. Experiments were run on equipment that is supported in part by the Data Analysis and Visualization Cyberinfrastructure funded by NSF under Grant OCI 0959097, as well as on equipment that is supported by the Cyberinfrastructure for Computational Research funded by NSF under Grant CNS 0821727.
Author Disclosure Statement
No competing financial interests exist.