
Abstract
Abstract
Assessing preferred relative rigid body position and orientation is important in the description of biomolecular structures (such as proteins) and their interactions. In this article, we extend and apply the “symmetrical parameterization,” which we recently introduced in the kinematics community, to address problems in structural biology. We also review parameterization methods that are widely used in structural biology to describe relative rigid body motions (in particular, orientations) as a basis for comparison. The new symmetrical parameterization is useful in describing the relative biomolecular rigid body motions, where the parameters are symmetrical in the sense that the subunits of a complex biomolecular structure are described in the same way for the corresponding motion and its inverse. The properties of this new parameterization, singularity analysis, and inverse kinematics are also investigated in more detail. Finally, parameterization is applied to real biomolecular structures and a potential application to structure modeling of symmetric macromolecules to show the efficacy of the symmetrical parameterization in the field of computational structural biology.
1. Introduction
S
In those parameterization methods, one needs to fix one molecule in the space (base frame) and describe the relative position and orientation of another molecule (moving frame) by using one of the methods. However, when one wants to switch the roles of base and moving frames (i.e., changing the view), then the conventional methods require a little bit of manipulation, which might be cumbersome in actual computations. Recently, a new method, called the symmetrical parameterization, has been developed where the corresponding parameters are free from changing the view between reference frames (Chirikjian, 2014; Kim and Chirikjian, 2015). The name “symmetrical parameterization” comes from the fact that the parameters are symmetrical in a given rigid body motion and its inverse. Hence, this new parameterization method has a high potential in computational structural biology, as well as in the robotic community.
This article builds on and is expanded from the prior work that was presented in the associated conference (Kim and Chirikjian, 2016). The organization of this article is as follows. First, we review the parameterization methods that have been popular in computational structural biology in Section 2. In particular, we emphasize the possibility of the symmetric description of a rigid body position and orientation. Then, in Section 3, we give an extended and updated analysis on the symmetrical parameterization. In particular, the updated definition and properties of the symmetrical parameterization are presented. In Section 4, the application of the symmetrical parameterization to relevant biological macromolecules is presented, where we describe how to extract corresponding symmetrical parameters, given the symmetric subunits, and discuss potential application of symmetrical parameterization to the calculation of energy functions, which is important in the process of structural prediction of symmetrical large biocomplexes. Finally, Section 5 concludes the article.
2. Classical Parameterization on the Rotations
Description of rotations and translations (i.e., orientations and positions) in terms of kinematics has become a standard in robotic and computational structural biology communities. A reader can refer to the in-depth classical treatment of rotations and rigid body motions (Angeles, 1988; Bottema and Roth, reprinted 1990; McCarthy, 1990; Selig, 2004, 2005; Chirikjian and Kyatkin, 2016). Here we review the classical methods to parameterize three-dimensional (3D) rotations, SO(3), that have been popular in molecular modeling (i.e., axis-angle parameters and Euler angles). These two parameterizations have been widely used in that the physical interpretations of the parameters are relatively straightforward to understand. Here we emphasize three-parameter descriptions rather than those that require four parameters and a constraint such as unit quaternions and Euler parameters. As is well known, any rotation in 3D space can be described by a 3 × 3 real matrix R satisfying the conditions
where
2.1. Euler angles
The Euler-angle parameterization starts from the consideration of counterclockwise rotations about the x = x1, y = x2, and z = x3 axes of a given coordinate system by the angle θ, which are given as
and
With these three fundamental matrices, Euler angles are defined by three such noncommutative rotations consecutively applied. For example, the ZXZ, ZYZ, and ZYX Euler angle parameterizations are defined as
Note that each of three fundamental rotations in Equations (1), (2), and (3) is symmetrical in the sense that
However, the ZYX Euler angles are not symmetrical since
If one wants to express this in the form of RZYX (α′, β′, γ′), then one finds that the resulting form, which requires a daunting task, would involve arc-trigonometric functions that are transcendental.
The most popular choices of Euler angles are the ZXZ and ZYZ angles. In both cases, the ranges of angles for these choices are 0 ≤ α ≤ 2π, 0 ≤ β ≤ π, and 0 ≤ γ ≤ 2π. One can find the relationships between the ZXZ and ZYZ Euler angles as follows. First when the ZYZ Euler angles are used, one can see that
hence, it follows that
Now let us investigate the possibility of the symmetric property in the inversion operation. One might think that the ZXZ Euler angles are symmetrical such as (α, β, γ) → (−γ, −β, −α) just because
However, it follows that the resulting angles are no longer in the correct range as mentioned earlier. Partial corrections in terms of matching the range can be done by changing −γ → 2π − γ and −α → 2π − α. Yet, how to put −β back into the correct range is not clear. Furthermore, as will be discussed later, this inversion formula turns out to be not consistent with the inverse kinematics of the corresponding Euler angles, which shows the complexity in terms of the inversion of this parameterization. Therefore, Euler angles do not have the symmetric property in terms of the corresponding inversion operation.
2.2. Axis-angle and exponential coordinates
The axis-angle parameterization results from the Rodrigues formula, which is written as
where
and the corresponding operation to construct N from
Regarding the ranges of the parameters, there are several choices. One can choose
This parameterization is almost symmetric in the sense that given
However, an issue arises when we consider the range of the parameters for
Again the similar convention is applied such that Φ = 0 and Φ = 2π always correspond to the same point, and all values of Φ map to the same point when Θ = 0 or π. Then, one can find that
which means that (θ, Φ, Θ) → (θ, Φ ± π, π − Θ). This obviously raises a problem that the correct value in ±signs must be checked to make sure that the parameters are in the correct range.
The above problem can be resolved when the Lie algebra so(3) (a set of all 3 × 3 real skew-symmetric matrices) is used. To be more specific, let
Here Ei (i = 1, 2, 3) denotes a skew-symmetric matrix corresponding to the ith natural basis vector
Then, as is well known in kinematics, exp X becomes a rotation matrix. These three parameters (x1, x2, x3) form the exponential coordinates. In fact, exp X can be interpreted as the rotation around axis
where X = θN. This parameterization is very convenient for many applications, as it is well behaved over the open ball of radius defined by
that is, the inversion operation is simply
2.3. Singularity analysis
The determinant of the Jacobian matrix corresponding to the parameters of interest is important in assessing the quality of parameterization. It is well known [see Chirikjian and Kyatkin (2016)] that given rotation with any three parameters
and the “left” Jacobian is
These are related to each other as
For the Euler angle parameterization, when we use the ZXZ Euler angles, then we have
and from Equation (9), it follows that
which indicates that the singularity occurs when β = 0 or π.
3. Symmetrical Parameterization on the Rotations and Rigid Body Motions
As we have seen in the previous sections, the classical parameterization methods do not possess the symmetric property in forward and inverse operations. In this section, a symmetrical parameterization is presented. The main purpose of a symmetrical parameterization is to parameterize rotations and rigid body motions in a “symmetrical” way, that is, given a rotation or a rigid body motion, the corresponding inverse looks the same way. Mathematically, given a 3 × 3 rotation matrix R(
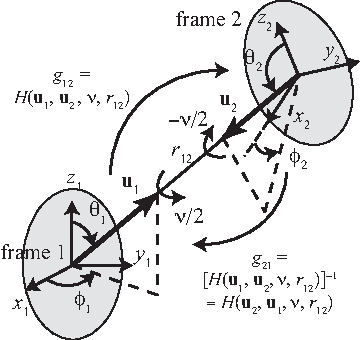
Symmetrical parameterization for a three-dimensional full rigid body motion.
3.1. Rotations in 3D
To define the symmetrical parameterization in SO(3), we need to define a special form of the rotation matrix R(
Note that θab should be defined as the unique angle in the range [0, π] such that
Some properties of the transference matrix R(
and
which can be shown from Equation (13) and the elementary properties of the cross product. Another important property is
where A∈SO(3) is an arbitrary rotation, and
Also, the following expression holds
for any angles α and β, because rot (
With the information so far, our symmetrical parameterization for rotations is defined as
As depicted in Figure 1, let
where
and
Physically, R1 represents the rotation half way starting at frame 1 and moving toward frame 2. And
Now let us consider the inverse as
In other words, inversion operation is simply written as
or
in its component form, which shows that parameterization is indeed symmetrical.
3.2. Rigid body motions in 3D
Suppose there are two rigid bodies, and reference frame 1 and 2 are attached to each body (Fig. 1). The position and orientation of the second body (frame 2) viewed from the first body (frame 1) are expressed as g12 = (R12, r12
Here the translation vector is described by the spherical coordinates (i.e.,
where we use the notations
On the other hand, the position and orientation of frame 1 (the first body) from frame 2 (the second body) become g21 = (R21, r12
Note that the vectors
from which one can see why the transference matrix in 3D rotation is defined in the form of R(
Finally, one can see that the inversion formula for these parameters is written as
or in the component form,
which shows that parameterization is indeed symmetrical.
3.3. Singularity analysis
In this section, we investigate the singularities of parameterization in Equation (18) when
and
where
and
with sθ = sin θ, cθ = cos θ, and vθ = 1 − cos θ.
Then, when we consider R(
Finally, it follows that
Note that |det J | does not depend on φ and v. This shows that as long as
3.4. Inverse kinematics
Inverse kinematics is especially useful when one wants to extract the symmetrical parameters, given rigid body motions of biomolecules. Given a homogeneous transformation matrix
(A∈SO(3) and
Then, by considering the fact that
Next, the rotational part is expressed as
Let T1 = trace(A−R(
Applying the half-tangent rule as
enables us to obtain the following equation
from which the angular parameter is calculated as
among which the value that falls into (0, 2π) is chosen so that v/2∈[0, π) (as long as
4. Application to Biomolecular Structures
In this section, we discuss the application of symmetrical parameterization to biomolecular structures. In the first subsection, we consider three molecules, a chaperon (GroEL/GroES complex), the Prohead domain of the HK97 viral capsid, and the structure of an actin filament. In these examples, the corresponding symmetrical parameters are obtained by considering a pair of subunits in the symmetric complex structures, through which we compare the symmetrical parameterization with other methods. In the subsequent subsection, we discuss a potential application of symmetrical parameterization to the calculation of energy scoring functions, which is one of the important steps in the process of structural prediction of symmetrical large biocomplexes (André et al., 2007; DiMaio et al., 2011).
4.1. Extracting the symmetrical parameters
First, let us consider a chaperon (GroEL/GroES complex), which is a large biomolecular complex that guides proper protein folding. X-ray crystallography has revealed the structure of chaperonin complex: it consists of several subunits, including GroEL, GroES, and seven bound adenosine diphosphate (ADP) molecules [Protein Data Bank (PDB) entry: 1AON] (Xu et al., 1997). In particular, we consider a ring structure made by seven GroEL subunits (cis-GroEL). See Figure 2a for the structure of cis-GroEL.
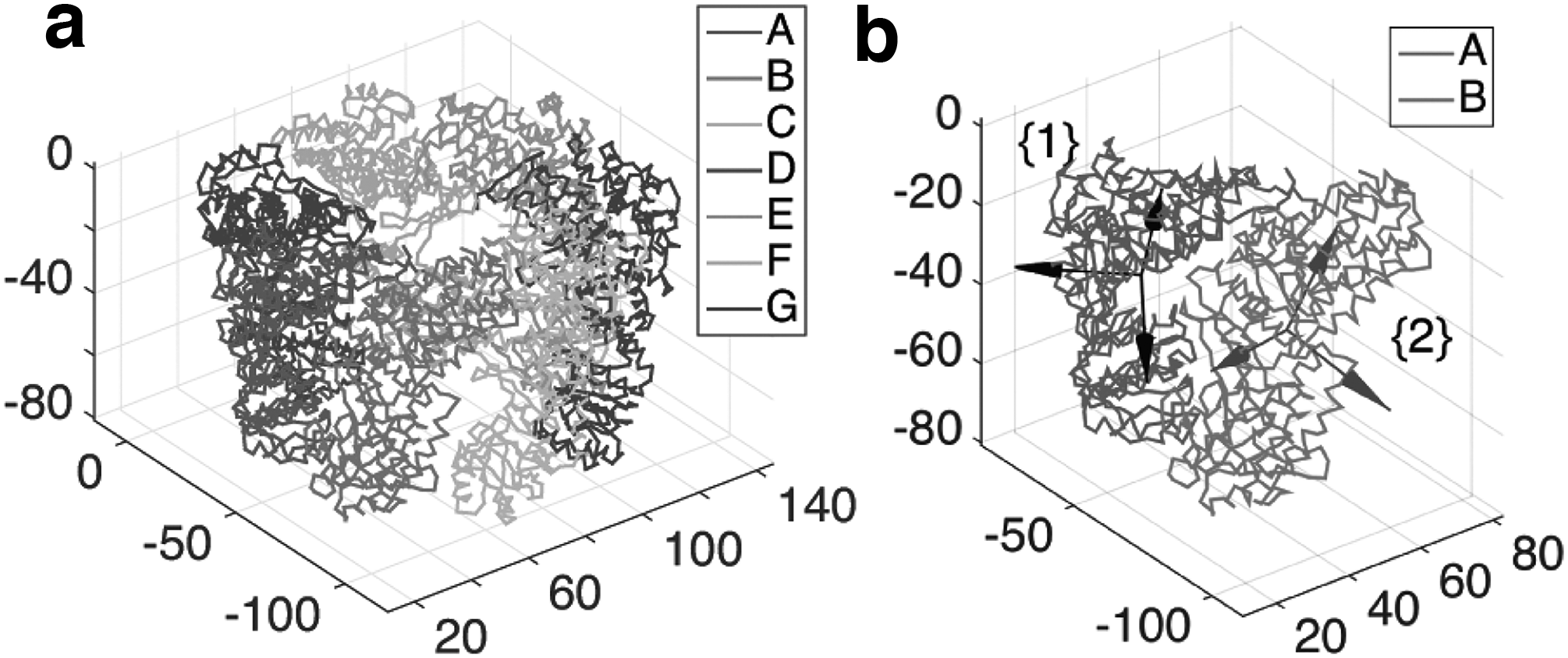
Let us consider the rigid body motion from chain A to chain B. As shown in Figure 2b, frame 1 and 2 are attached to the center of mass of chain A and B, respectively. Frame 1 and 2 are obtained by the method of standardizing the coordinates (Kim et al., 2002). The relative rigid body motion from frame 1 to frame 2 is described by the following homogeneous transformation matrix
For the comparison, we calculate the ZXZ Euler angles and axis-angle parameters for the rotation between frame 1 and 2. First, the ZXZ Euler angles are calculated by using the following formula. Given R = (Rij)∈SO(3) (i, j = 1, 2, 3), angles are computed as
which gives
For the inverse orientation, we obtain
Hence, the ZXZ Euler angles are not symmetrical. Also, in comparison with a possible inversion formula as discussed earlier, this shows the complication of finding parameters between forward and inverse orientations. If we consider the axis-angle parameters, then we obtain
for the orientation from frame 1 and frame 2, and
for the inverse orientation. Note that
Now let us consider the symmetrical parameterization shown in Equations (18) and (20). By applying the inverse kinematics in Section 3.4, we obtain
Since these parameters are symmetrical, this set is complete in the sense that it also determines the inverse motion.
Next, we consider the structure of the HK97 viral capsid (Wikoff et al., 2000; Conway et al., 2001). We take the Prohead II domain (PDB entry: 1IF0), which consists of a hexamer (chain A to F) and a part of a pentamer (chain G) (Conway et al., 2001). Here we consider a hexamer assembly as shown in Figure 3a. As in the previous example, we select chain A and B to describe the relative motion from A to B (Fig. 3b). The relative rigid body motion is described by the homogeneous transformation matrix as
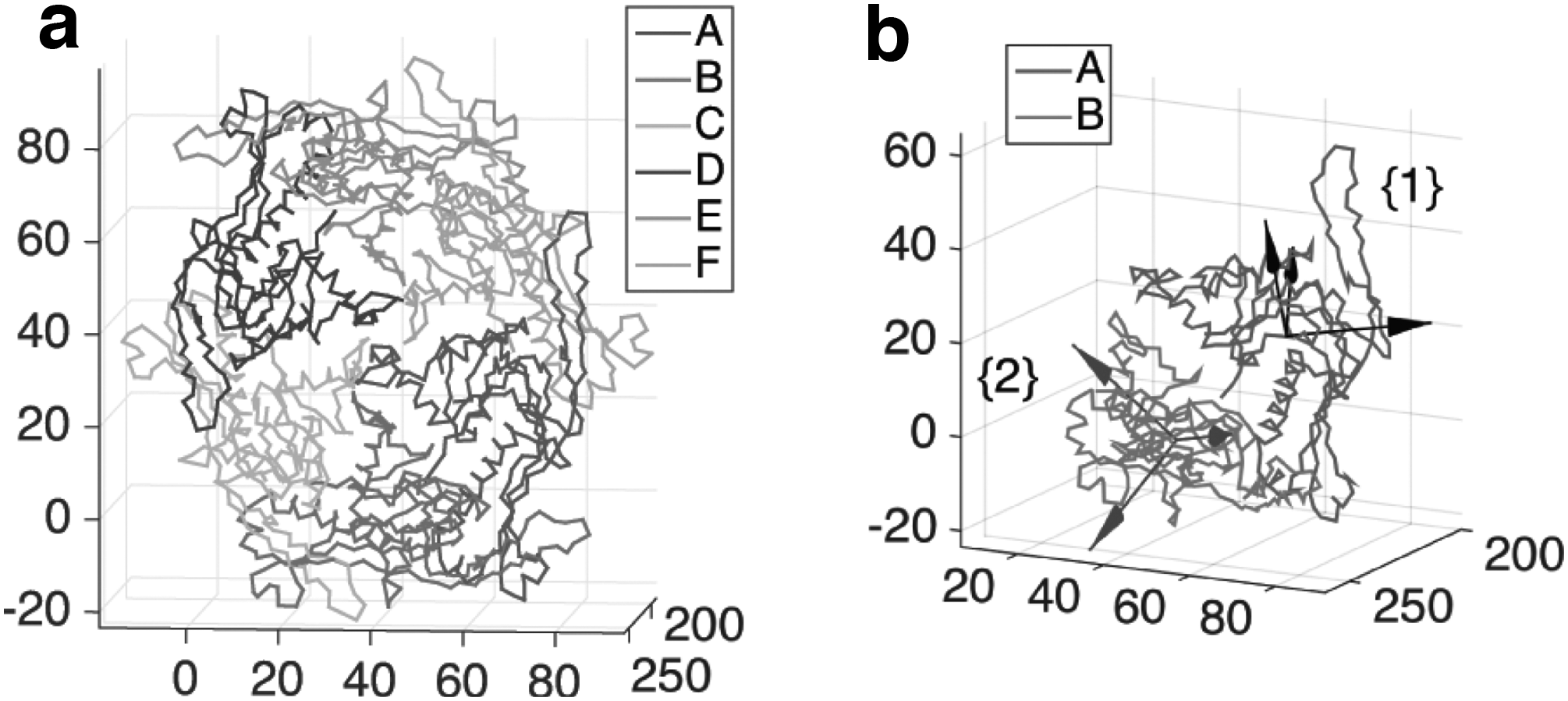
The ZXZ Euler angles for the rotation are obtained as
For the inverse rotation, we obtain
Also, the axis-angle parameters for the rotation are obtained as
and for the inverse rotation, we obtain
which all together show the complexity of the parameters between forward and inverse rotations.
The symmetrical parameters are obtained as
and these parameters are all that need to determine the inverse motion.
Finally, let us consider the structure of an actin filament (PDB entry: 3G37, see Fig. 4a), which has been revealed through the cryo-EM method (Murakami et al., 2010). Actin filaments are important in many cellular functions such as cell motility and maintenance of cell cortex. This is an example of helical symmetries where, unlike the previous two examples, a rigid body motion (i.e., rotation and translation) along a single axis is enough to represent the structure. Here we consider two subunits as in Figure 4b. The relative rigid body motion is described by the homogeneous transformation matrix as
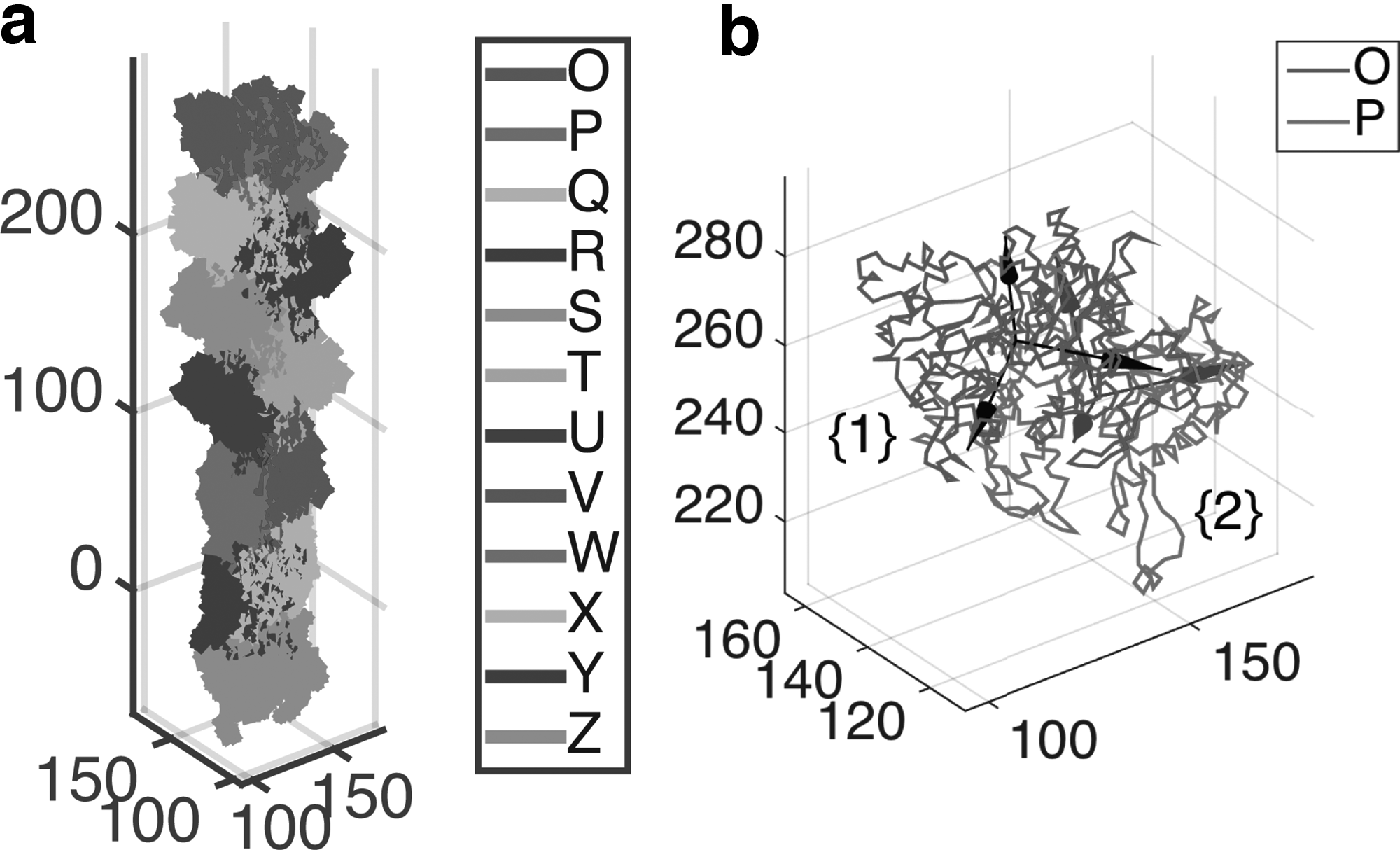
The ZXZ Euler angles for the rotation are obtained as
For the inverse rotation, we obtain
Also the axis-angle parameters for the rotation are obtained as
and for the inverse rotation, we obtain
which again all together show the complexity of calculating the parameters between forward and inverse rotations. However, the symmetrical parameters are obtained as
which again shows that this set of parameters is what we need to describe forward and inverse rigid body transformations between two subunits.
This as a whole emphasizes the usefulness of symmetrical parameterization in describing the structures of relative positions and orientations between subunits in complex biomolecules regardless of the symmetry type.
4.2. Calculating energy functions between symmetric subunits
In this section, we discuss a potential advantage of symmetrical parameterization on structure determination of symmetric large biomolecules.
In modeling symmetric large macromolecules, due to the structural symmetry, one does not need to consider the whole subunits, rather one subunit and the relative rigid body transformation between subunits are often enough. In practice, one subunit, called the “master subunit,” is often enough for the structure prediction (DiMaio et al., 2011). This master subunit is copied and pasted according to the symmetry to generate the entire symmetric complex structure. In doing so, apparently, attaching a reference frame at each copy of the master subunit has a big advantage. This is particularly true when considering interactions between subunits, which is important in predicting the structure of a large protein assembly. Instead of considering all the individual atom coordinates, one can use the parameters for the rigid body transformation between subunits in computing energy scoring functions between subunits. Another advantage of using a reference frame to each subunit is that it can facilitate the computation of the gradient of the energy function during structure prediction process than when to use the coordinates of a single subunit and the information on symmetry (André et al., 2007).
In calculating the energy function of a symmetric large complex, as well as interactions between atoms/residues inside each subunit, one also has to consider interactions between subunits. Due to the symmetry, however, one does not need to consider all the pairs. Rather pairs generated between the master subunit and the adjacent subunits (called the “slave subunits”), which are copied from the master subunit, are usually enough (DiMaio et al., 2011). There are many kinds of energy functions included in the protein structure prediction. For example, Rosetta, a software that is widely used in structure prediction, includes more than 15 energy scoring functions (Rohl et al., 2004). Many functions involve interactions between residues, secondary structural motifs, and so on. Hence, to explain how symmetrical parameterization contributes to the modeling of symmetric large macromolecules, let us consider a function of the distance between residues between subunits, which can represent the energy scoring functions such as Lennard-Jones potential energy, electric interactions, steric repulsion, and hydrogen bonding.
Let
denotes the distance between the i-th residue in the master subunit (
denotes the distance between the i-th residue in the master subunit (
where dm denotes the distance at the minimum potential, and
where H denotes the homogeneous transformation matrix of gm,1 with given symmetrical parameters. Then, the j-th atom or residue in the copy 1 subunit from the reference frame attached to the master subunit can be simply computed as
or
where · denotes the action of
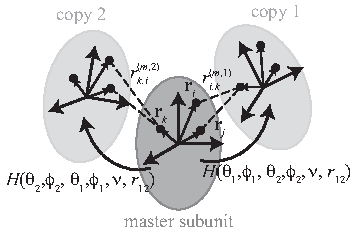
A schematic that illustrates the concept of how to consider interactions between the master subunit and two adjacent slave subunits (copy 1 and 2).
Next, we want to compute the second term,
If one utilizes other parameterizations (such as Euler angles for the orientation and the spherical coordinates for the translation), then as we have seen in the previous section, we have to calculate the corresponding parameters for the inverse transformation. However, when we use symmetrical parameterization, we can simply perform the following:
which means that we do not need to compute the corresponding parameters for the inverse rigid body transformation. Rather, we can still use the same set of symmetrical parameters for that purpose.
To illustrate this, Figure 6 shows two conformations of three subunits in GroEL (red and magenta, respectively, denote the master subunit and slave subunits), given two different sets of symmetrical parameters. The symmetrical parameters are as follows:
for Figure 6a, and
for Figure 6b.
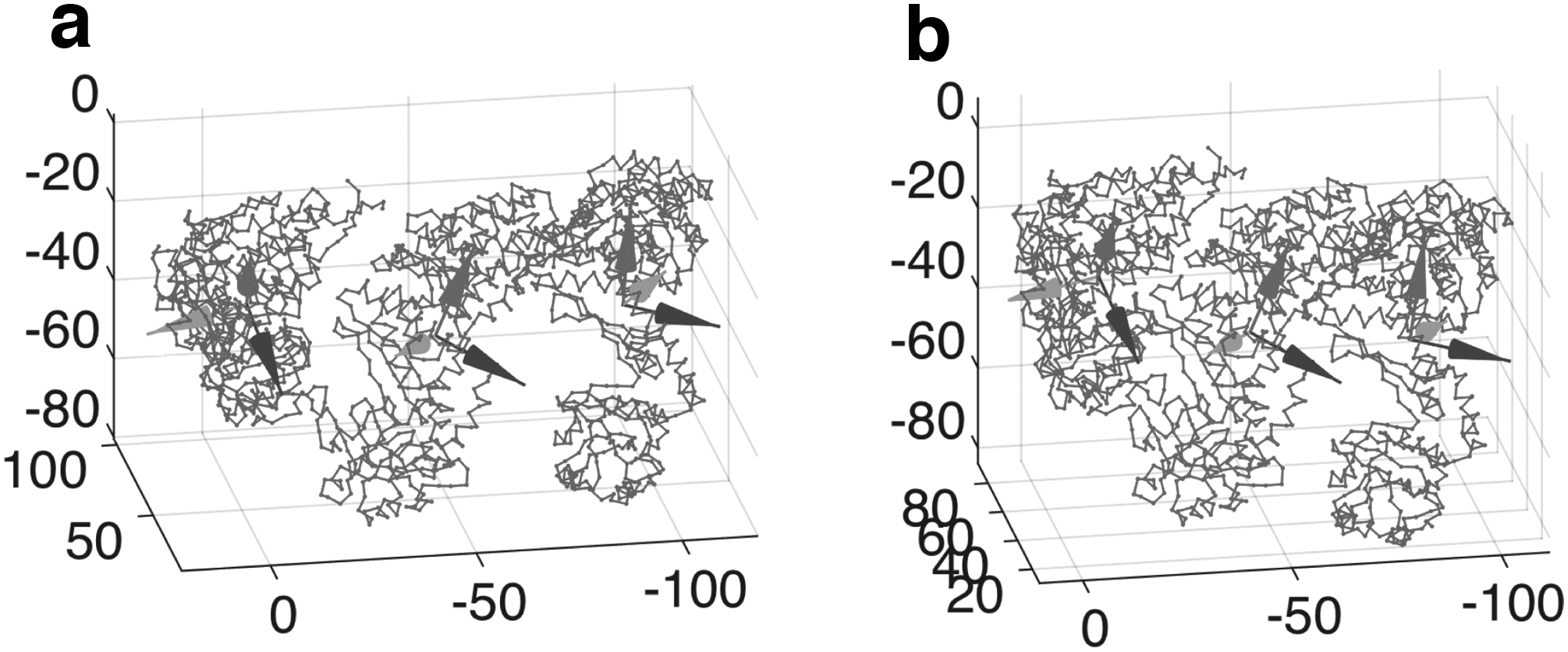
Two conformations of three subunits in GroEL structure. The red-colored subunit denotes the master subunit, whereas the magenta-colored ones (copy 1 and 2) denote the slave subunits, which are copied and pasted from the master subunit by a rigid body transformation.
For comparison, let us apply other parameterizations. For the rotation, we use the ZXZ Euler angles (α, β, and γ) and axis-angle parameters (θ, Φ, and Θ). For the translation, we use spherical coordinates (r, θ, and φ). First, let us consider the configuration in Figure 6a. The rigid body transformation from the master unit to copy 1 (i.e., gm,1) has
On the contrary, the inverse rigid body transformation (i.e., gm,2) has
Next, let us consider Figure 6b. gm,1 has the following parameters:
while the corresponding inverse gm,2 has
This clearly illustrates that, given a set of symmetrical parameters, we can describe the conformation of three adjacent subunits very efficiently.
Finally, using the symmetrical parameterization, the energy function becomes the function of the symmetrical parameters, together with a set of atom/residue coordinates of the master subunit
For a detailed discussion, let us consider the Lennard-Jones potential energy again. Suppose there are N residues in the master subunit. Total potential energy from the interactions between the master subunit and slave subunits can be written, assuming that constant parameters (ɛ and dm) are the same regardless of pairs of residues, as
where
On the contrary, to compute
and hence one can write
As we have seen in the previous examples, the relationships between the parameters of the forward and inverse rigid body transformations are complicated in any of the classical parameterization methods. However, if one uses the symmetrical parameterization method, then one can write
This is of a great advantage when it comes to computing energy scoring functions. All together, this demonstrates that using symmetrical parameterization has a high potential in the structure prediction of a large symmetric macromolecule.
5. Conclusions
In this article, we presented an extension of our previous analysis of symmetrical parameterization for rotations and rigid body motions in 3D space, and applied this parameterization to model spatial relationships between rigid subunits in biomolecular structures and complexes. To compare with current alternatives, we presented a review of the most common parameterizations of 3D rotations (axis-angle parameters and Euler angles) used in the structural biology field. Our new parameterization has a symmetric property such that the inversion of a given rigid body rotation and translation is described by the same set of parameters used in describing the original motion. Singularity analysis for parameterization on 3D rotations was presented, and an inverse kinematic procedure for a given homogeneous transformation matrix followed accordingly. Finally, we applied symmetrical parameterization to a couple of biological macromolecules that consist of several subunits, and discussed its potential application to structure prediction of symmetric macromolecules, which emphasizes the efficacy of presented symmetrical parameterization on the structural description of large complex biological macromolecules.
Footnotes
Acknowledgments
This work was supported by NSF grants (award numbers: CCF-1640970 and IIS-1619050) and the National Institute of General Medical Sciences of the NIH (award number: R01GM113240).
Author Disclosure Statement
No competing financial interests exist.