
Abstract
Abstract
This article introduces a multiscale framework for spatiotemporal modeling of protein–protein interaction. Cellular protein molecules represent multivalent species that contain modular features, such as binding domains and phosphorylation motifs. The binding and transformations of these features occur at a small time and spatial scale. On the other hand, space and time involved in protein diffusion, colocalization, and formation of complexes could be relatively large. Here, we present an agent-based framework integrated with a multiscale Brownian Dynamics (BD) simulation algorithm. The framework employs spatial graphs to describe multivalent molecules and complexes with their site-specific details. By implementing a time-adaptive feature, the BD algorithm enables efficient computation while capturing the site-specific interactions of the diffusing species at the sub-nanometer scale. We demonstrate these capabilities by modeling two multivalent molecules, one representing a ligand and the other a receptor, in a two-dimensional plane (cell membrane). Using the model, we show that the algorithm can accelerate computation by orders of magnitudes in both concentrated and dilute regimes. We also show that the algorithm enables robust model predictions against a wide range of selection of time step sizes.
1. Introduction
A
Several models have been developed in the past to study multivalent antigen-mediated IgE receptor clustering in the plasma membrane of mast cells (Goldstein and Perelson, 1984; Hlavacek et al., 1999; Monine et al., 2010; Mahajan et al., 2014). However, these models were either equation-based deterministic models or developed by using a nonspatial Kinetic Monte Carlo approach called network-free simulation (Yang et al., 2008; Sneddon et al., 2011; Yang and Hlavacek, 2011). The explicit geometric features of molecules and complexes were missing in these models.
In this article, we introduce a multiscale Brownian Dynamics (BD) simulation algorithm that enables accelerated computation while capturing the sub-nanometer scale site-specific interactions of the molecules and their complexes. We develop a model of a trivalent ligand and a trivalent receptor molecule and their diffusion, rotation, and site-specific binding in the plasma membrane of a cell. The multivalent molecules and their site-specific features are described by spatial graphs. The mobile ligand-receptor graphs form larger graphs (multimolecular complexes) that evolve dynamically during the course of a simulation. The lateral and rotational motion of the molecules and evolving complexes are simulated by using the BD algorithm. Using the model, we provide analysis on the computational performance and accuracy of our algorithm. We show that the algorithm speeds up computation by orders of magnitude without compromising the accuracy in both concentrated and dilute regimes. Moreover, we show that the time-adaptive feature enables robust predictions against a wide range of selection of time steps.
2. Methods
Later, we first provide details of the model. We then explain the multiscale BD algorithm.
2.1. Model
The model is a reaction-diffusion model of multivalent ligand-receptor interaction in the cell plasma membrane. The model is developed by using the agent-based approach (Gilbert, 2008). More details are provided later.
2.1.1. Cell membrane, molecules, and complexes
We consider a 1 μm2 two-dimensional plane, which represents a small part of the cell membrane (∼0.3% of the surface area of a spherical cell of 5 μm radius). We consider a trivalent ligand and a trivalent receptor molecule that we describe by spatial graphs (Grünert and Dittrich, 2010) (Fig. 1). A spatial graph is a collection of small circles (nodes). The spatial organization of these nodes creates a coarse-grained structure of a molecule. When two or more such molecule graphs combine in a prescribed manner, they form a complex graph. Each ligand or receptor molecule contains three equally spaced binding arms. In each binding arm, one node is designated as the reaction center (Fig. 1). The reaction centers serve as the mutual recognition sites of the ligand and receptor molecules. In a complex, the molecules remain linked via these reaction centers (Fig. 1C).
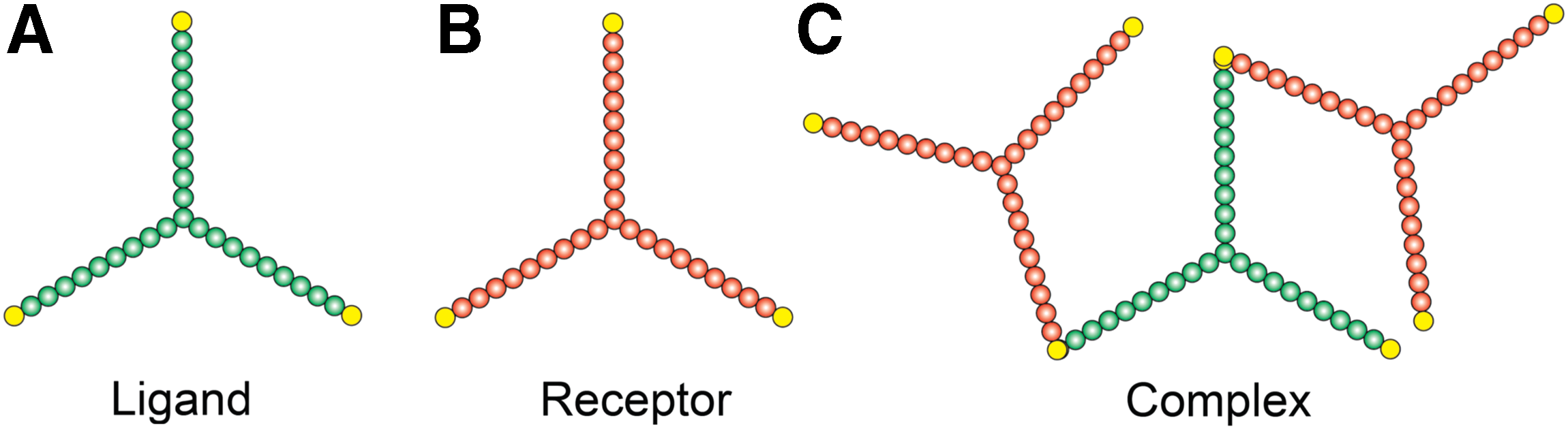
Illustration of the species (molecules and their complexes).
2.1.2. Motion of molecules and complexes
We consider both lateral diffusion (translation) and rotation of the molecule and complex graphs. The diffusion and rotation rates are determined by their effective size. To define the effective size for a graph, we consider a circle that completely contains the corresponding structure (Fig. 2A). The circle is centered at the center of mass of the structure. The center of mass is determined by the mass of individual nodes and their spatial locations. We consider identical mass for all nodes because they have an identical size in the model (Table 1). The radius of the circle extends from this center to the farthest node. We define the circle area as the effective region (A), and its radius as the effective size (a). For a molecule graph, a remains fixed over time. However, for a complex graph, a is designated as
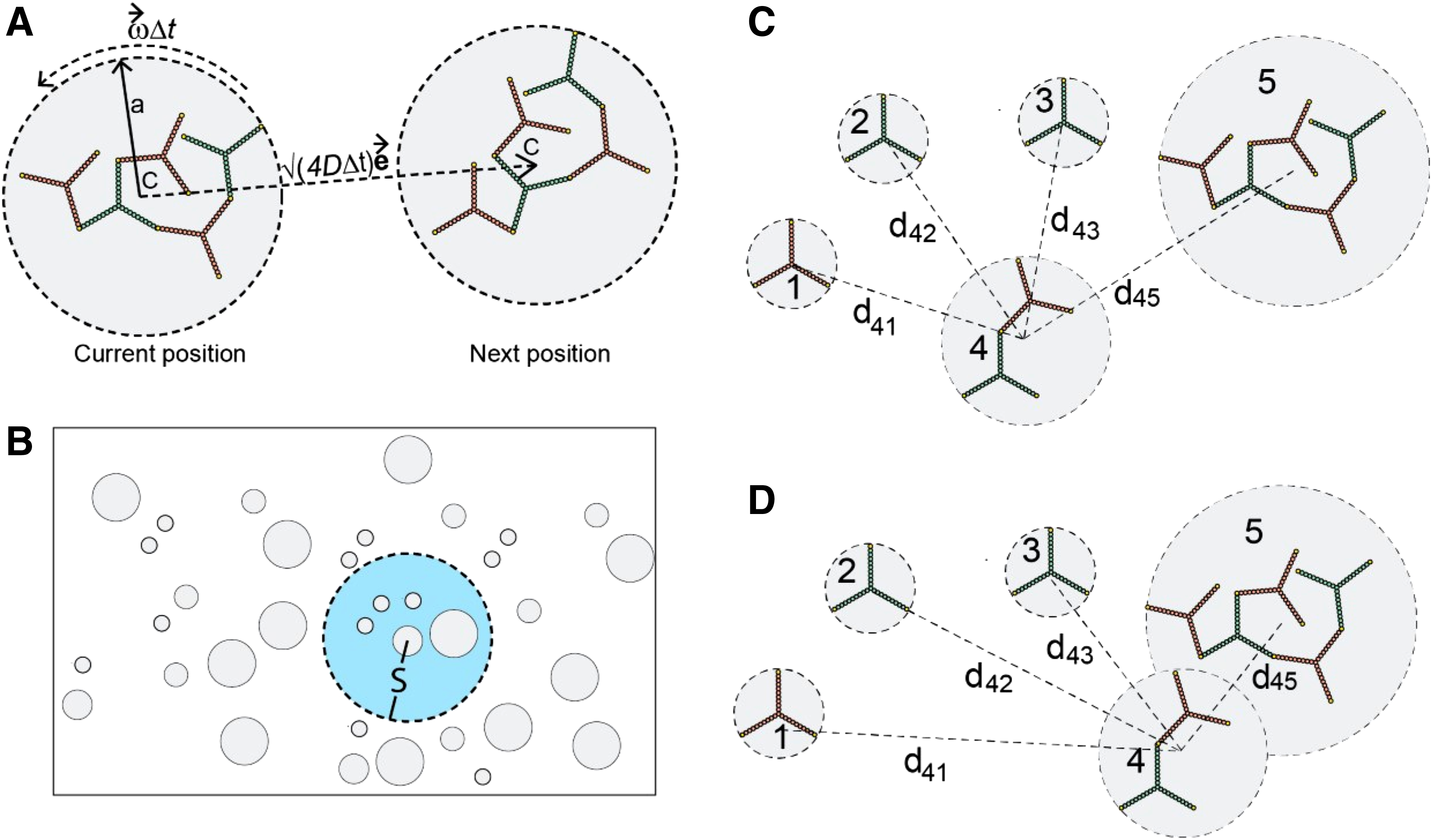
Illustration of the BD simulation algorithm.
For a ligand or receptor molecule, we predefine their diffusion constants Dm (Table 1). For a complex graph, diffusion evolves based on the following rule:
During the course of a simulation, we translate or rotate each graph as a rigid body. In each time step
2.2. Time-adaptive BD algorithm
The multiscale simulation algorithm is illustrated in Figure 2. The algorithm ensures collision-free motion of the structures defined by the molecule and complex graphs, as discussed later.
2.2.1. Calculation of adaptive step size
For each molecule or complex agent, the algorithm assigns a local search domain of radius S. In Figure 2B, the two concentric gray and blue circles illustrate a species agent and its search domain, respectively. The gray circle at the center represents the effective region of the species. The blue circle represents the search domain whose radius (S) is a tunable parameter.
In the algorithm, an upper bound
In each BD step, the algorithm evaluates
If
If
2.2.2. Collision rejection and site-specific binding
The algorithm allows an overlap of two circles (effective graph regions) while ensuring that the graphs within those circles are advanced in a collision-free manner. If a move results in a collision between two graphs, the move is rejected for the two graphs only. We consider a thin reaction layer,
2.2.3. Code implementation
The simulation algorithm and the model were specified in C++. The C++ code is provided in the Supplementary Material. All simulations were conducted on a dedicated node on AMAZON Elastic Compute Cloud (EC2): c3.4xlarge (16vCPU, 30 GiB,
3. Results
3.1. Simulation of multivalent ligand-receptor systems
In Figure 3A and B, we show snapshots from our simulations. At time zero, the molecules were initiated at random nonoverlapping positions in the membrane domain, and snapshots were taken after 500 seconds of simulation.
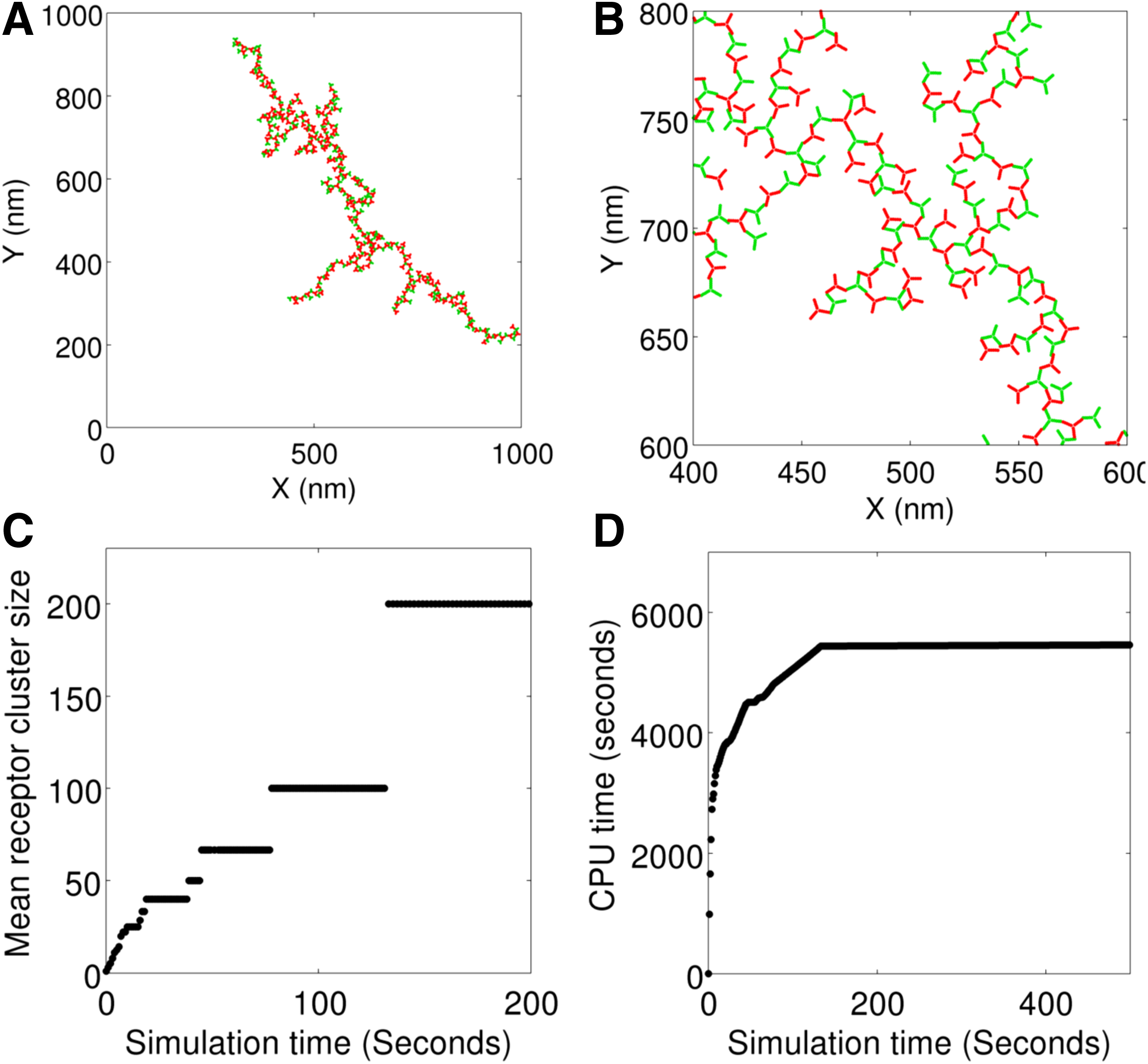
Simulation of a trivalent ligand-trivalent receptor system. The ligand and receptor molecules are indicated in green and red, respectively.
Figure 3C shows the time evolution of receptor clustering. The mean cluster size represents the average of the number of receptors incorporated in the ligand-receptor complexes at the indicated time points. Initially, all receptors were available as isolated molecules, and, hence, the mean cluster size was 1 at time zero. As time progressed, larger (and fewer) clusters populated in the system. Figure 3D shows how the computation time (CPU time) evolved as the simulation progressed. Most of the computation was spent in the first few seconds because the system was in a concentrated regime (species were either single molecules or small complexes). As the simulation progressed, larger (and fewer) species formed and the system approached a dilute regime. In the dilute regime, the adaptive algorithm took larger steps, thus accelerating computation.
Figure 4 shows some representative snapshots from simulations that involved different other multivalent ligand-receptor combinations. A movie file (movie.mp4) is provided in the Supplementary Material showing the time evolution of a trivalent ligand–trivalent receptor system.
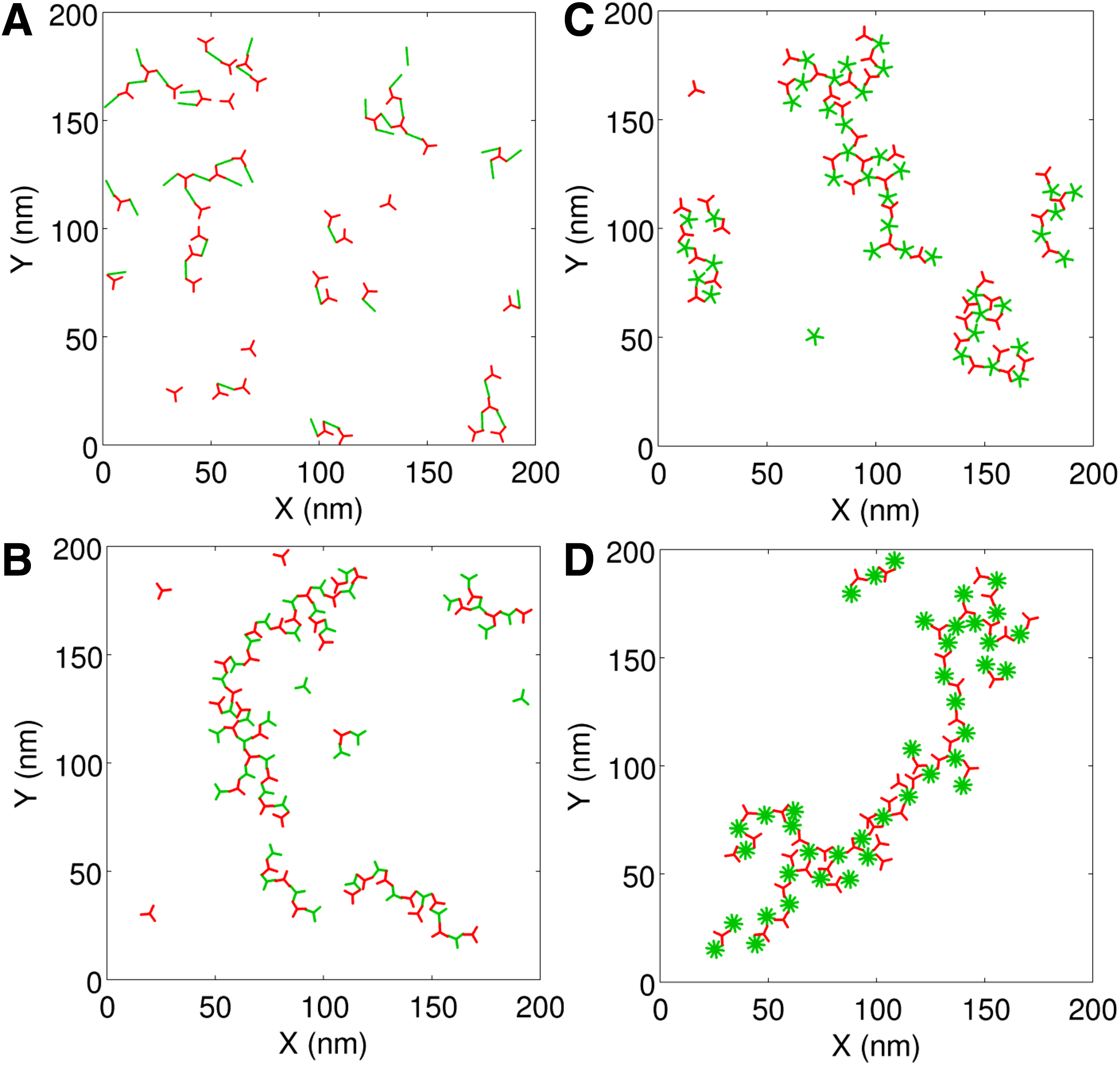
Simulation snapshots of multivalent ligand (green)-receptor (red) systems. In all cases, the receptor is trivalent whereas the ligand is bivalent
3.2. Gain in computation speed
We evaluated the computational performance of our algorithm against a naive implementation of the model (Fig. 5). In the naive implementation, the time step

Computational speed gain in the time-adaptive algorithm. All simulations involved 100 trivalent receptors and 100 trivalent ligand molecules.
The figure shows a dramatic reduction in computation time in the adaptive algorithm. The difference between the naive and adaptive algorithm is even more pronounced in the dilute regime, where the naive algorithm spent most of the computation in particle advancement.
3.3. Robustness and accuracy
The results we have discussed so far represent
We investigated three different combinations of
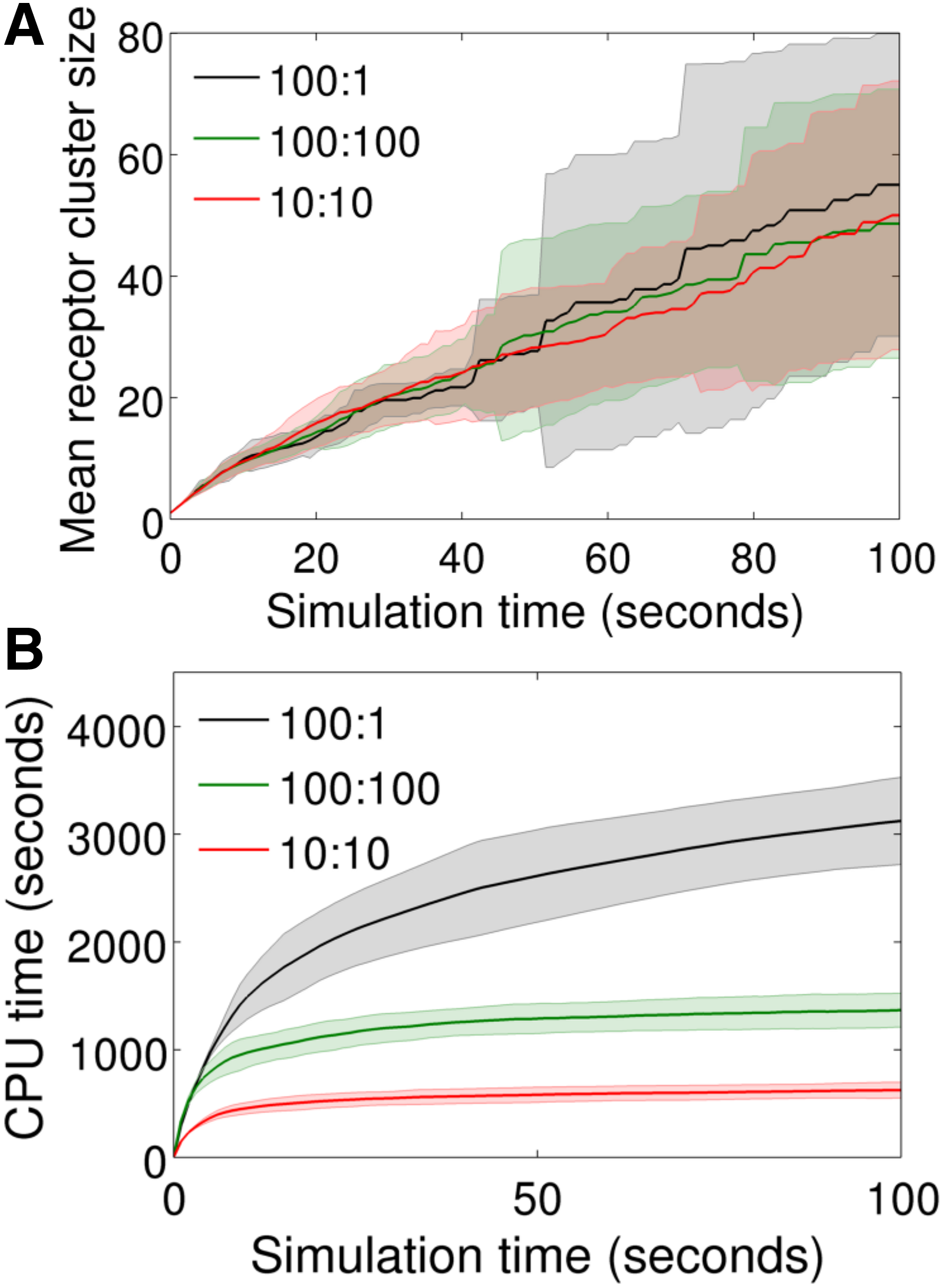
Sensitivity and computational performance under different combinations of S and
Figure 6A shows the predicted mean receptor cluster size as a function of time under the three conditions mentioned earlier. Except for the stochastic variations, the predictions are closely similar in all three cases, indicating the robust behavior of the time adaptive feature. Despite the distinct upper bounds (
Figure 6B compares computation time among the three conditions mentioned earlier. First, it is interesting to note that the 100:1 case took only ∼3500 CPU seconds despite the adaptive feature being compromised with
The two other cases (100:100 and 10:10) revealed modest improvements in computation speed compared with the 100:1 case. Nevertheless, these two cases plateaued rapidly as the system approached from the concentrated to dilute regime. This indicates that the adaptive feature could be of greater advantage in the dilute regimes. On the other hand, the local search feature alone can improve computation speed significantly in a concentrated regime.
4. Discussion
In this work, we have demonstrated a computationally efficient algorithm for modeling reaction-diffusion systems involving multivalent molecules. Here, we limited our focus to modeling cell-surface ligand-receptor assembly, a key early step of immunoreceptor and receptor tyrosine kinase signaling (Schlessinger, 2000; Puffer et al., 2007; Deak et al., 2016). The approach and analysis could be adapted for modeling and simulation of more complex multivalent species or particle interactions in molecular biology or other fields.
Among the existing spatiotemporal modeling tools, SRSim (Grünert and Dittrich, 2010) provides a unique capability for spatiotemporal modeling of macromolecular assembly. SRSim also employs spatial graphs to represent protein molecules and complexes. However, making SRSim simulations more comparable could be expensive for the time and spatial scales of signaling protein interactions. MCell (Bartol et al., 2015) is a spatiotemporal modeling tool with the special capability to consider complex geometries for cellular compartments. However, MCell treats reaction species as featureless points (Kerr et al., 2008). Therefore, it has a limited ability to incorporate site-specific attributes of the biomolecules. VirtualCell (Loew and Schaff, 2001) is another spatiotemporal modeling tool. However, it is primarily limited to partial differential equation-based deterministic modeling. It might be useful to extend our multiscale approach to incorporate some of the unique features of the tools just mentioned. In particular, our algorithm might be coupled to features creating complex geometries for the reaction compartments, as implemented in MCell.
In this work, we have limited the scope to modeling protein–protein interactions in the plasma membrane, which is often treated as a two-dimensional space. However, a three-dimensional extension of the approach might enable modeling the cytoplasmic and extracellular compartments. We expect that the computational benefits of our algorithm will be significant in a three-dimensional space where the simulation of particle advancement could be more expensive than that in a two-dimensional system.
Footnotes
Acknowledgments
This work was supported by the National Science Foundation (1609642). No additional external funding was received to support this work. The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of this article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.