
Abstract
Abstract
The advent of next-generation sequencing (NGS) technologies has revolutionized the world of genomic research. Millions of sequences are generated in a short period of time and they provide intriguing insights to the researcher. Many NGS platforms have evolved over a period of time and their efficiency has been ever increasing. Still, primarily because of the chemistry, glitch in the sequencing machine and human handling errors, some artifacts tend to exist in the final sequence data set. These sequence errors have a profound impact on the downstream analyses and may provide misleading information. Hence, filtering of these erroneous reads has become inevitable and myriad of tools are available for this purpose. However, many of them are accessible as a command line interface that requires the user to enter each command manually. Here, we report EasyQC, a tool for NGS data quality control (QC) with a graphical user interface providing options to carry out trimming of NGS reads based on quality, length, homopolymer, and ambiguous bases. EasyQC also possesses features such as format converter, paired end merger, adapter trimmer, and a graph generator that generates quality distribution, length distribution, GC content, and base composition graphs. Comparison of raw and processed sequence data sets using EasyQC suggested significant increase in overall quality of the sequences. Testing of EasyQC using NGS data sets on a standalone desktop proved to be relatively faster. EasyQC is developed using PERL modules and can be executed in Windows and Linux platforms. With the various QC features, easy interface for end users, and cross-platform compatibility, EasyQC would be a valuable addition to the already existing tools facilitating better downstream analyses.
1. Introduction
E
Bioinformatic analyses remain to be the vital and complex part of any NGS project, involving a plethora of heterogeneous sequence data (Pabinger et al., 2014). Considering the wide array of applications possessed by NGS, it becomes essential to be wary of the nature of data obtained from them. Artifacts such as low-quality reads and adapter contamination are commonly observed in NGS data. Presence of low quality sequences shall severely impact the downstream analyses, viz, assembly, annotation, SNP detection, etc. (Dai et al., 2010). There are multiple factors affecting the quality of reads across various NGS platforms and each one of them has their own quality control (QC) pipelines.
Despite the primary filtering of sequences, there exists a need to carry out an elaborate QC process to ensure that only high-quality sequences are carried further for subsequent analyses. Earlier reports (Gloor et al., 2010; Caporaso et al., 2011) suggest that there was a loss of 40–70% reads after the initial quality filtering of raw sequences. Many tools have come into existence since then for NGS data QC, such as NGS QC toolkit (Dai et al., 2010), FastQC (Andrews, 2010), ClinQC (Pandey et al., 2016), PRINSEQ (Schmieder and Edwards, 2011), FAST-X Toolkit (Gordon, 2010), Galaxy (Goecks et al., 2010), Tag Cleaner (Schmieder et al., 2010), and QC-Chain (Zhou et al., 2013). Few of them exist as online tools and others are standalone tools with their own dependencies and limitations. These dependencies may at times hinder data processing and not many tools have user interface to carry out seamless operations.
In this study, we have developed EasyQC, an integrated standalone tool for NGS data QC with interactive user interface and cross-platform compatibility. EasyQC facilitates the end user to prepare NGS data in FASTQ format for secondary analyses through QC pipeline and other associated features. Considering the ever growing need of infallible tool for NGS data QC, we believe that EasyQC will be of significant utility to the research fraternity.
2. Development and Implementation
All the scripts of EasyQC have been written using PERL programming language (PERL v5.22). Perl Tk package has been implemented for developing the user interface. The functionalities of EasyQC have been segregated into various modules that ensure better maintainability of the tool. The workflow of EasyQC is described in Figure 1. The user interface of the various utilities of EasyQC is depicted in Figure 2. Graphical outputs were generated using GD::Graphs and URI::Google Chart packages. Functioning of the tool has been exhaustively tested in Windows and Linux (Ubuntu v16.04) environments.

The workflow of EasyQC pipeline.

User interface of EasyQC containing multiple tabs enabling the user to execute required process.
3. Results and Discussion
EasyQC is an open source desktop tool with cross-operating system compatibility, facilitating the QC process of huge NGS data sets with great accuracy. EasyQC includes various modules such as format converter, adapter trimmer, and QC graph generator apart from the pipeline that carries out filtering based on length, quality, ambiguous bases, and homopolymer repeats as well. The key features of EasyQC are described hereunder.
3.1. EasyQC features and pipeline
3.1.1. Format converter
This module of EasyQC mediates the conversion of. fastq files to. fasta files. It verifies the accuracy of the input FASTQ file and converts it to FASTA file upon successful format check.
3.1.2. Adapter trimmer
Adapter or primer trimming is a vital step in NGS data QC. It is important to remove the forward and reverse adapter sequences from the data set. The adapter trimmer module of EasyQC takes the forward and reverse adapter sequences as input and trims the sequences accordingly.
3.1.3. Paired end merging
Many sequencing protocols that are in use today sequence the DNA fragments from both ends. Merging of these paired end reads is important in genome assembly and other analyses. The paired end merging module of EasyQC uses FLASH (Magoč and Salzberg, 2011) to merge the input FASTQ files.
3.1.4. Graph generator
This feature of EasyQC enables the user to obtain graphical outputs of length distribution, average quality distribution, ATGC composition, and GC content of the input sequence data set.
3.1.5. Quality-based trimming
Removal of low quality sequences is essential, since they can have a significant impact on the downstream analyses of NGS data. This module of EasyQC gets the quality threshold for each base along with the percentage threshold for each sequence as input. Sequences that contain lesser percentage of high-quality bases than the threshold will be removed from the data set.
3.1.6. Length-based trimming
At times, data sets possess sequences with a very wide range of read lengths and it is important to remove those sequences that fall short of the required length. This module trims those sequences that are shorter than the input threshold length value.
3.1.7. Ambiguous-based trimming
Presence of ambiguous bases has significant impact on the subsequent alignment process. This feature fetches the threshold percentage of the ambiguous bases that shall be permitted in a sequence and those with a greater percentage will be removed.
3.1.8. Homopolymer trimming
Homopolymer repeats in a sequence shall also cause misalignment of the sequences leading to false results. This module takes the cutoff value for the homopolymers to be present in a sequence and those that exceed this cutoff value will be taken off from the data set.
The trimming modules are designed as a single pipeline in EasyQC. The user shall upload the input sequence data set and specify the threshold values of all the modules at the same time and execute the pipeline.
3.2. Input and output of EasyQC
EasyQC takes FASTQ files generated from any NGS platform as input for all its modules. The outputs from EasyQC are as hereunder.
3.2.1. Paired end merging
The output FASTQ file containing the stitched paired end sequences based on user input using FLASH gets stored in a new folder with the statistics of the number of sequences that were merged.
3.2.2. QC statistics
The QC pipeline of EasyQC provides the statistics of the total sequences that got filtered in each step of the QC processes based on the filter parameters provided by the user (Fig. 3). The output sequences in FASTQ and FASTA format are generated at every step of the pipeline. These sequences can directly be used for downstream analyses. The comparison of quality of raw sequence data set with that processed by EasyQC with a quality cutoff of Q30 was carried out using FastQC, and the results indicated significant increase in the overall quality of the reads (Fig. 4).
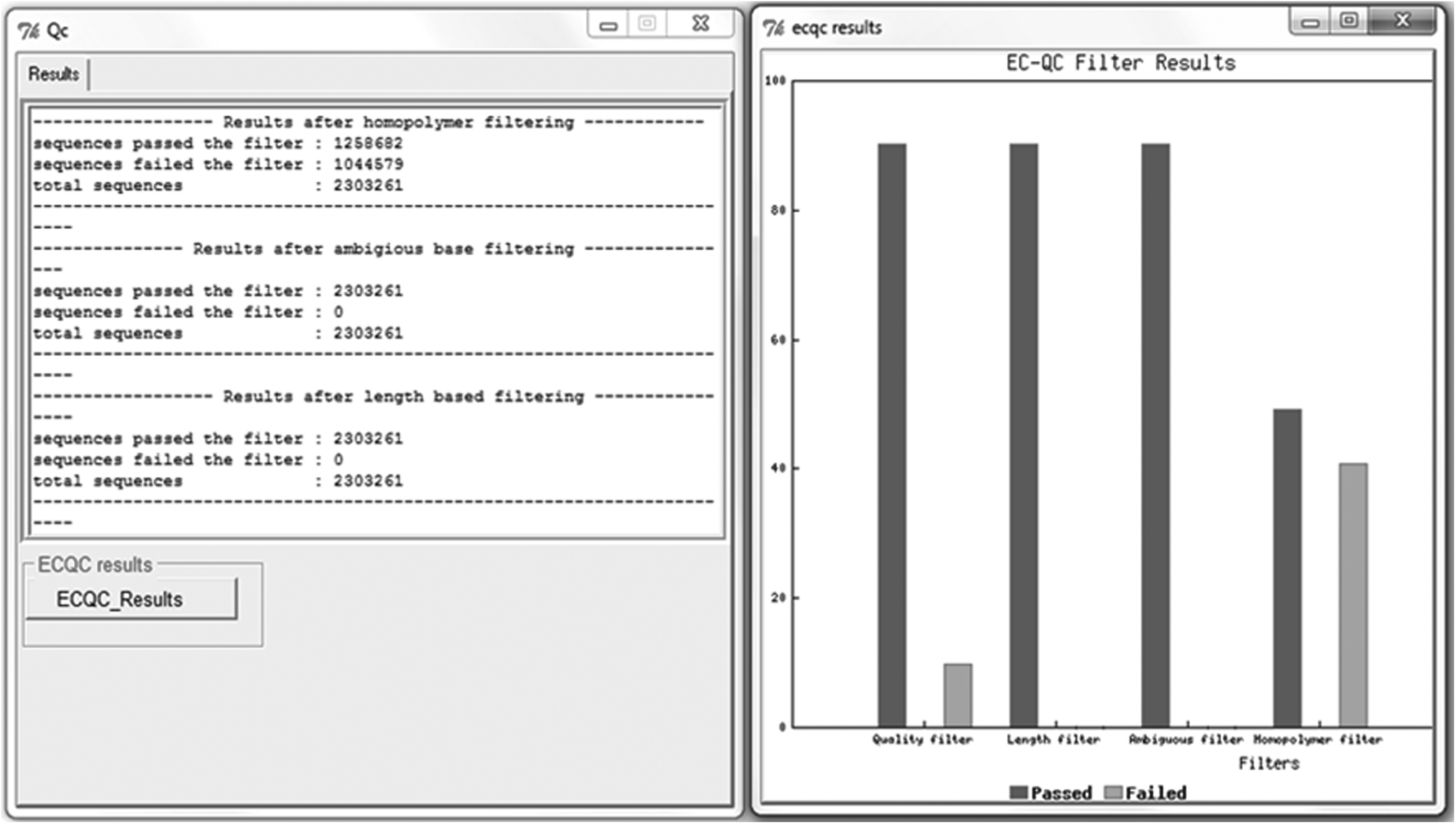
Results of the trimming pipeline in EasyQC. The graph represents the number of sequences filtered at each stage of the trimming process.
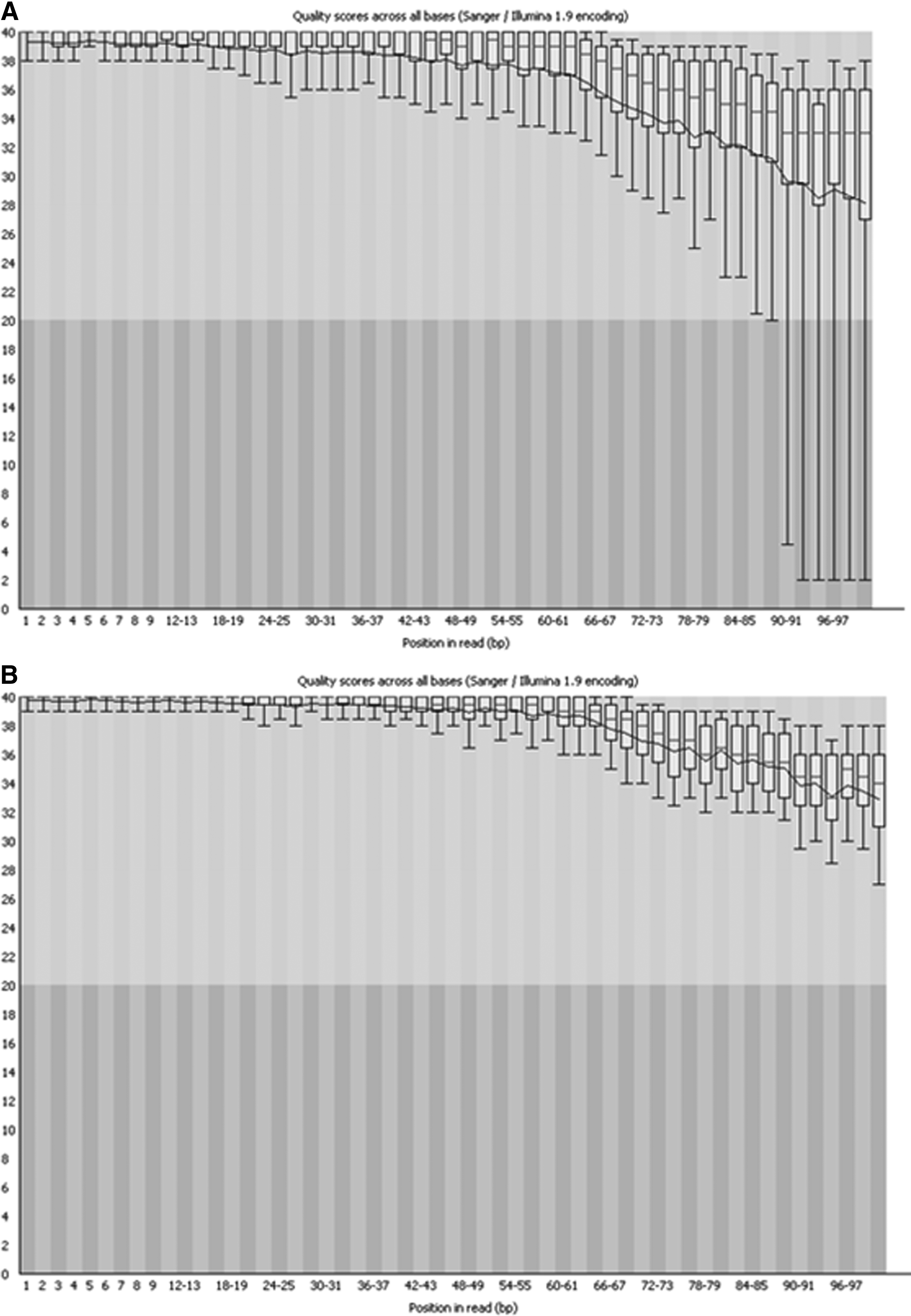
Comparison of per base quality of
3.2.3. Graphical output
The graphs module generates four different graphs as described earlier (Fig. 5) and is stored in a separate folder in the same location from where the input files were uploaded.
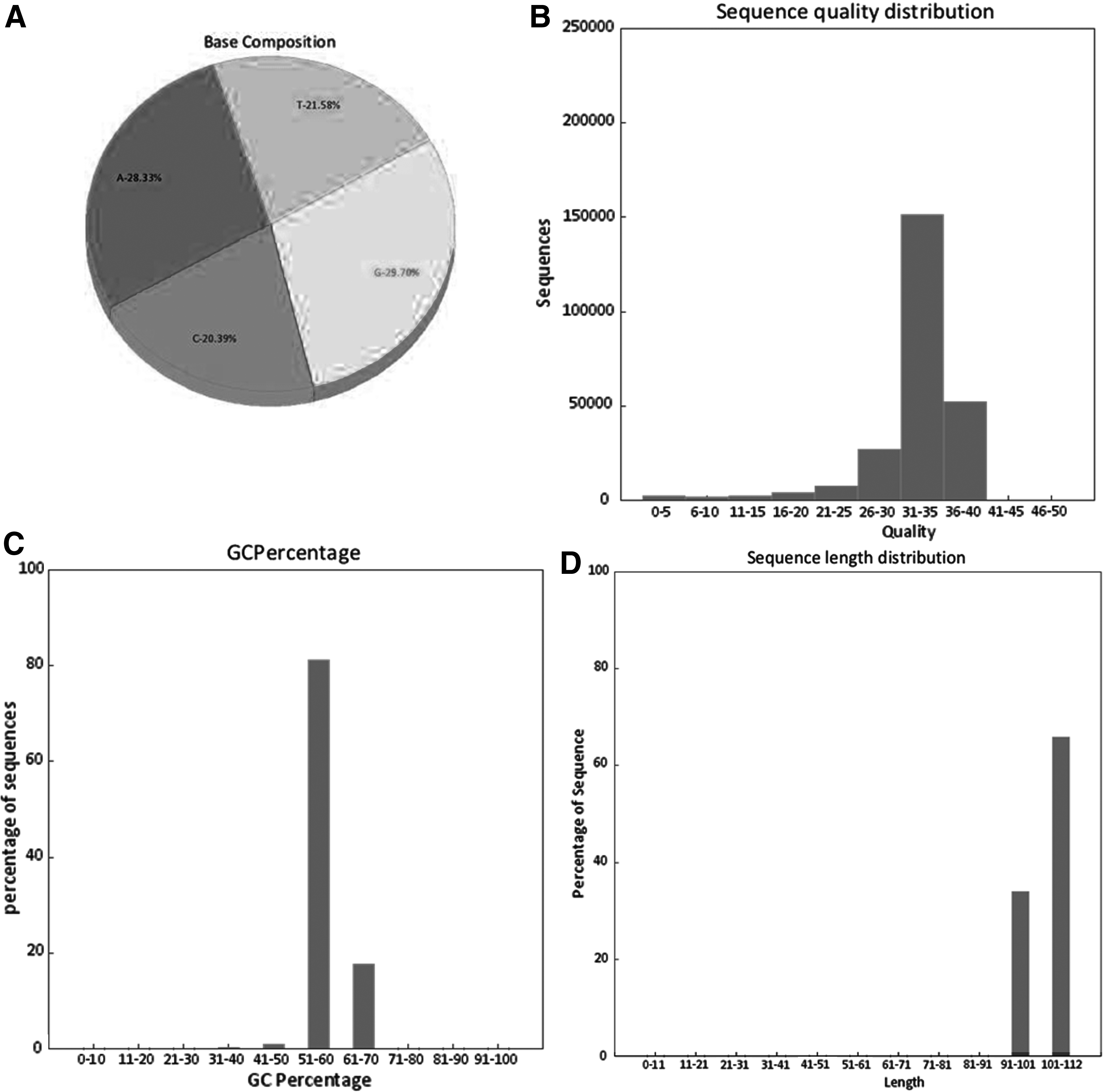
Graphical output of an NGS data set obtained from EasyQC.
3.3. Comparison with existing tools
The comparison of various features available in EasyQC with that of other QC tools is described in Table 1. Although many toolkits are available, not many exist as desktop standalones with a user interface. EasyQC provides that interactive facility to the users through its interface with a flexibility to work across Windows and Linux platforms.
Comparison of Various Features in EasyQC with Other Quality Control Tools
3.4. Performance evaluation
The performance of EasyQC was validated using various Illumina sequence data sets containing sequences ranging from 0.3 to 2.8 million. These evaluations were carried out on a standalone Dell precision T7600 workstation with Intel Xeon(R) E5-2560 processor and 32GB RAM (operating system: Windows 7). The time taken for the execution of QC pipeline for various sequence data sets is given in Table 2. Comparison of EasyQC with other tools such as NGSQC and PRINSEQ for filtering of sequences based on quality score and length was carried out using a data set containing 2.3 million sequences. This was carried out in HP Z620 Workstation with Intel(R) Xeon(R) CPU E5-2620 v2 processor and 32GB RAM (operating system: Ubuntu v16.04). The quality cutoff was set at Q30, whereas the length threshold was set to 100 bp.
Execution Time of the Quality Control Pipeline of EasyQC with Next-Generation Sequencing Data Sets
In addition, the percentage of high-quality bases per sequence was set at 90 (which is the default value) in EasyQC. Comparison of the post-QC data sets (Fig. 6) indicated that the filtering of sequences by EasyQC was more stringent and there were no significant deviation in the quality distribution as well. The time taken for the QC process was 4 minutes 28 seconds for EasyQC, whereas it took 1 minute 20 seconds for NGSQC and 1 minute 40 seconds for PRINSEQ. The relatively higher time taken by EasyQC can be attributed to the execution of percentage of high-quality base filter module, which is not present in the other two tools, in addition to the overall quality filter option. Furthermore, EasyQC provides the output data set containing the sequences that passed and failed in each step of the QC process that adds to its execution time, which is not provided by the tools compared herewith.

Comparison of post-QC evaluation of sample data set using EasyQC
4. Availability and installation
EasyQC is an open source tool available for free download at (www.niot.res.in/EasyQC). Separate installer files are available for Windows and Linux platforms. Steps to be followed while installing EasyQC and the installation of the dependent PERL packages are given in detail in the readme file available along with the installer.
5. Conclusion
With an interactive user interface, QC pipeline, and other associated features with cross-platform compatibility, EasyQC would be a significant addition to the already existing QC tools providing enhanced downstream analyses.
Footnotes
Acknowledgments
The authors gratefully acknowledge the financial support provided by the Ministry of Earth Sciences, Government of India. The authors are thankful to Dr. M.A. Atmarand, Director, National Institute of Ocean Technology, for constant suggestions and encouragement to carry out this work.
Author Disclosure Statement
The authors declare that no competing financial interests exist.