
Abstract
Abstract
Nearly half of the human genome is made up of transposable elements (TEs), and there is evidence that TEs are involved in gene regulation. In this study, we have integrated publicly available genomic, epigenetic, and transcriptomic data to investigate this in a genome-wide manner. A bootstrapping statistical method was applied to minimize confounder effects from different repeat types. Our results show that although most TE classes are primarily associated with reduced gene expression, Alu elements are associated with upregulated gene expression. Furthermore, Alu elements had the highest probability of any TE class contributing to regulatory regions of any type defined by chromatin state. This suggests a general model where clade-specific short interspersed elements (SINEs) may contribute more to gene regulation than ancient/ancestral TEs. Our exhaustive analysis has extended and updated our understanding of TEs in terms of their global impact on gene regulation and suggests that the most recently derived types of TEs, that is, clade- or species-specific SINES, have the greatest overall impact on gene regulation.
1. Introduction
R
A number of existing studies have shown that TEs can influence host genes by providing novel promoters, splice sites, or post-transcriptional modifications to rewire different developmental regulatory and transcriptional networks (Kunarso et al., 2010; Lynch et al., 2011; Cowley and Oakey, 2013). TEs tend to regulate gene expression through several mechanisms (van de Lagemaat et al., 2003; Kunarso et al., 2010; Cowley and Oakey, 2013). For example, L1 elements show a stronger negative correlation with expression levels than the gene length (Jjingo et al., 2011), and the presence of L1 sequences within genes can lower transcriptional activity (Han et al., 2004). Past studies have found that TEs have contributed to nearly half of the active regulatory elements (REs) of the human genome (Jacques et al., 2013) by altering gene promoters and creating alternative promoters and enhancers to regulate gene activity (Medstrand et al., 2001; Conley et al., 2008; Franchini et al., 2011). Moreover, 60% of TEs in both humans and mice were located in intronic regions and all TE families in humans and mice can exonize, supporting the view that TEs may create new genes and exons by promoting the formation of novel or alternative transcripts (Piriyapongsa et al., 2007; Sela et al., 2007).
In this study, TEs in the human genome were analyzed using genome-wide datasets associated with gene regulation. These datasets enabled an assessment of the association of TEs with REs based on chromatin states for the first time, as marked by histone modification within six human cell lines and gene ontology (GO) enrichment, as well as overall transcriptome profiles.
Although, a number of studies have analyzed the association between TEs and gene regulation in the human genome, they did not consider whether gene intervals contained multiple repeat types or not. This can cause a confounding factor when attempting to determine the regulatory impact of a specific repeat type. In our study, we eliminated confounding effects by focusing on gene intervals that only contained one type of repeat and used a weighted bootstrap approach to minimize any influence of co-occurring elements. Our method presents a novel way to investigate the association of a specific TE type with gene expression.
2. Methods
2.1. The distribution of repetitive elements in the human genome
To assess how human TEs were distributed in genes, we compared different genic regions containing TEs. Based on Repbase (hg19, see detail in Supplementary Note—Section 1.1) (Jurka et al., 2005; Kohany et al., 2006) annotations identified by RepeatMasker (www.repeatmasker.org), repeat elements in humans were divided into two categories: human/primate-specific repeats and repeats shared with different species (Supplementary Table S1). Intergenic regions, exons and introns within 5′UTR, CDS, and 3′UTR regions from RefSeq genes (Bellone et al., 2013) were then compared with these different categories of repeats by using BEDTools (Quinlan and Hall, 2010). Next, we generated the summarized distributions of repetitive elements overlapping these regions by calculating the proportions of bases belonging to repetitive elements within each of the combined sets of regions:
The code repository for the above can be found at https://github.com/UofABioinformaticsHub/RepeatElements
2.2. The occurrence of transposable elements within regulatory regions
We further explored the association between any TE and the REs (Supplementary Note—Section 1.2) by calculating the proportion of nucleotides within each of the five sets of genic regions that were part of an RE for each of the six human cell lines. The proportion of nucleotides that were TEs within each RE was also calculated for each genic region. All proportions were subsequently transformed using the logit function for model fitting across tissues (Supplementary Table S2) using the following model:
where
A specific TE analysis was then performed using six of the major human TE classes based on the Repbase classification system (Alu, L1, L2, LTR, MIR, and DNA transposons). L1 elements were further resolved into either ancestral (L1M, L1PB, and L1PA subfamilies) or recent/clade specific (L1HS subfamily) based on their Repbase annotations. The proportions of nucleotides within each TE type that were also REs were calculated giving tissue-specific estimates of
2.3. The weighted bootstrap procedure for assessing the effects of a transposable element in each genic region
Many genes contain multiple TEs, with only a minority of genes containing a single TE (see detail in Supplementary Figs. S1 and S2 and Tables 1 and 2). To assess any effects on transcription due to the presence of a single TE, a weighted bootstrap approach was devised. For a given TE within each genic region within each individual tissue, the frequencies of co-occurring TEs and combinations of TEs were noted. Uniform sampling probabilities were then used for the set of genes containing a specific TE in a specific region, while sampling weights were assigned to genes lacking the specific TE based on TE composition, such that the TE content of the sampled set of reference genes matched that of the test set of genes based on defined categories. The mean difference in expression level, as measured by log2(TPM) (Supplementary Note—Section 1.3 and 1.4), and the difference in the proportions of genes detected as expressed were then used as variables of interest in the bootstrap procedure. The bootstrap was performed on sets of 1000 genes for 10,000 iterations using genic regions as defined above. When comparing expression levels, genes with zero read counts were omitted before bootstrapping.
To compensate for multiple testing considerations, confidence intervals were obtained across the
3. Results
3.1. Distribution of repetitive elements in the human genome
Initially, we investigated the distribution of repetitive elements in the human genome. We found that many repetitive elements overlapped with gene models from the human RefSeq gene datasets, and their distributions with respect to components of gene models are shown in Figure 1.

Distribution of repetitive elements overlapping with different human gene regions. Gene regions are shown on the x-axis, and the y-axis shows the percentages of genomic regions containing repetitive elements. Human-specific repeats were those annotated with Homo sapiens or primates as their origin (see Supplementary Table S1 for the list of human-specific repeat classes). The remaining repetitive regions were categorized as shared repeats. Fewer repetitive elements were found in coding exons and noncoding exons such as 5′ and 3′ UTRs. This indicates that repeat accumulation in these regions is selected against these regions in order to preserve regulatory and coding functions.
3.2. Transposable elements and gene regulation by chromatin states
To look for enrichment of TEs within REs, we looked at proportions of nucleotides with a TE in six defined REs as they appear in different components of the gene model. This represents the probability of a given nucleotide within an RE being from a TE, that is, p(TE|RE) (see Section 2) across the set of genic regions (Fig. 2A). One major observation from Figure 2A is that TEs have higher p(TE|RE) in insulators, while active promoters have the lowest repeat level in regulatory regions. The proportion of TEs in REs is lower than the genome-wide average based on p(TE|RE) values, with exceptions being most weak enhancers in noncoding regions. Unsurprisingly, CDS exons have the lowest repeat content. The overall under-representation of TEs in REs is likely indicative of negative selection for TEs in regulatory regions.

Analysis of the co-occurrence of TEs and REs across multiple genomic regions.
Pairwise comparisons of p(TE|RE) between different gene model compartments are shown in Figure 2B to identify significant differences (see Section 2) in repeat content between regulatory regions. Results show that TEs are more sparsely distributed across REs within CDS exons and 3′UTR than REs in other genic regions. In contrast, polycomb-repressed regions and insulators in intergenic regions were enriched for TEs in comparison with REs in other gene model compartments.
3.3. Different classes of transposable elements and their associations with chromatin state
A similar method was applied to characterize the distribution of REs within specific TE classes, using the probability of a nucleotide also belonging to a TE element p(TE|RE) (see Section 2). The most significant observation from Figure 3A was that Alu had the highest p(TE|RE) across all regulatory regions, while recently inserted L1 elements were least likely to be within REs compared with the other five repeat classes. Interestingly, MIR and L2, ancient repeats that have been inactive since before the mammalian radiation, are present at very low levels in the human genome and they have a relatively high p(TE|RE) in strong enhancer regions.
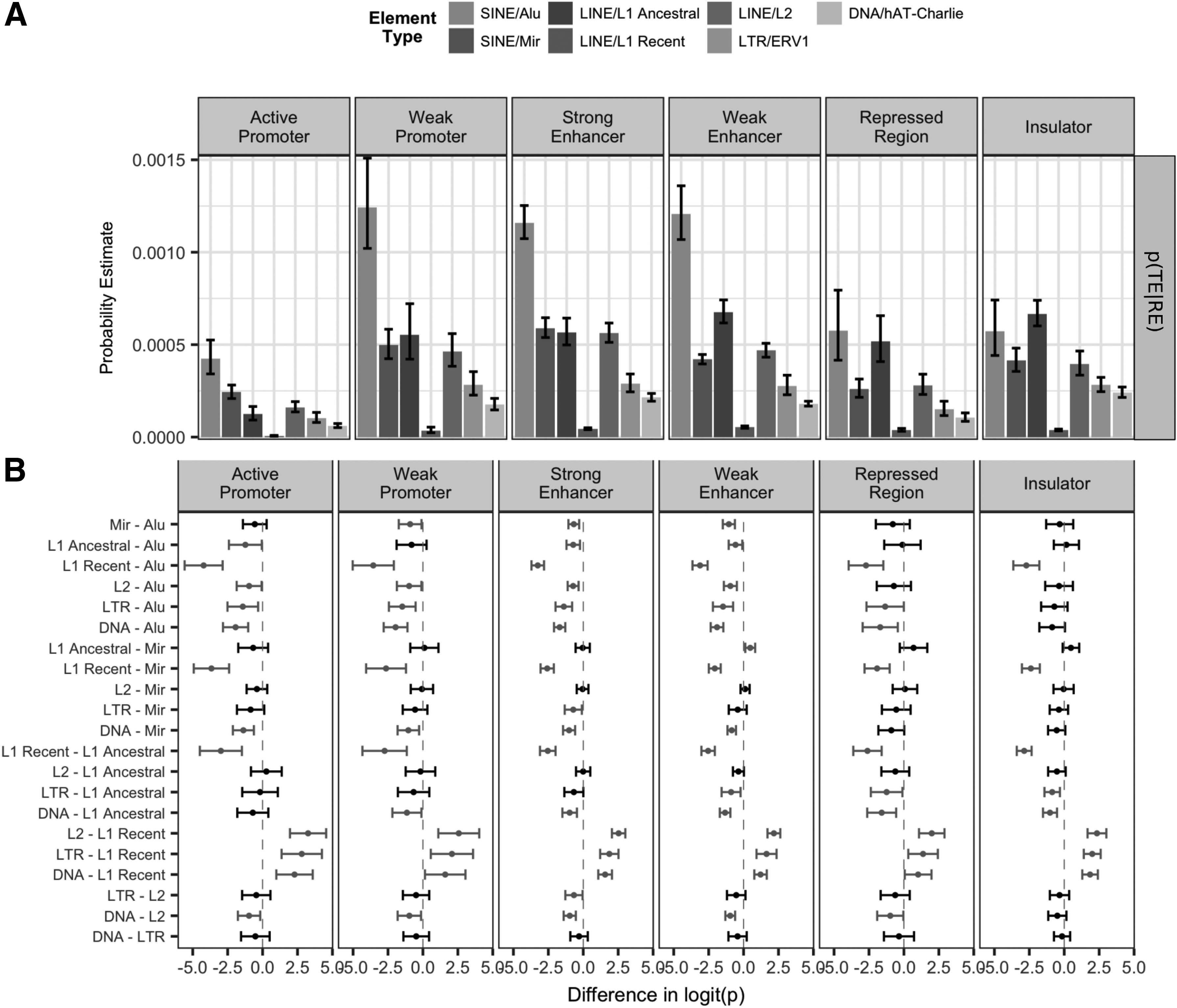
Analysis of the occurrence of REs within specific classes of TEs.
To identify differences within specific repeat types between regulatory regions, pairwise comparisons of p(TE|RE) were implemented and shown in Figure 3B (see Section 2). Among six defined regulatory regions, Alu elements contributed more to these regions compared with the other five repeat types. Ancestral L2 and MIR were found to be retained more than recent L1s across all REs. Another indication of potential exaptation of ancestral repeats was the observation that the recently inserted L1 had the lowest probability of being in an RE compared with other repeat types (Fig. 3B). However, LTR elements and DNA transposons were also more likely to be retained than recent L1s, suggesting some degree of exaptation for those repeats as well.
3.4. Effects on the probability of a gene being detected as expressed due to the presence of a transposable element across different gene model compartments
As chromatin states are not always indicative of changes in transcriptional activity, we further investigated any effects on human gene expression due to the presence of specific TE classes within each of the three genic regions.
To exclude confounder effects caused by multiple repeat types on the expression of single genes, the weighted bootstrap method was applied to both the probability of a gene being detected as expressed (Fig. 4A) and to the overall expression levels for those genes detected as expressed (Fig. 4B). This revealed that Alu elements are commonly associated with a higher probability of expression when located in 5′/3′ UTR regions across the majority of tissues (Fig. 4), while the presence of L1 elements in the proximal promoter showed a negative impact on the probability of a gene being detected as expressed in three of the six tissues, with the remaining tissues being directionally consistent and quite likely to be Type II errors (Supplementary Fig. S3A).
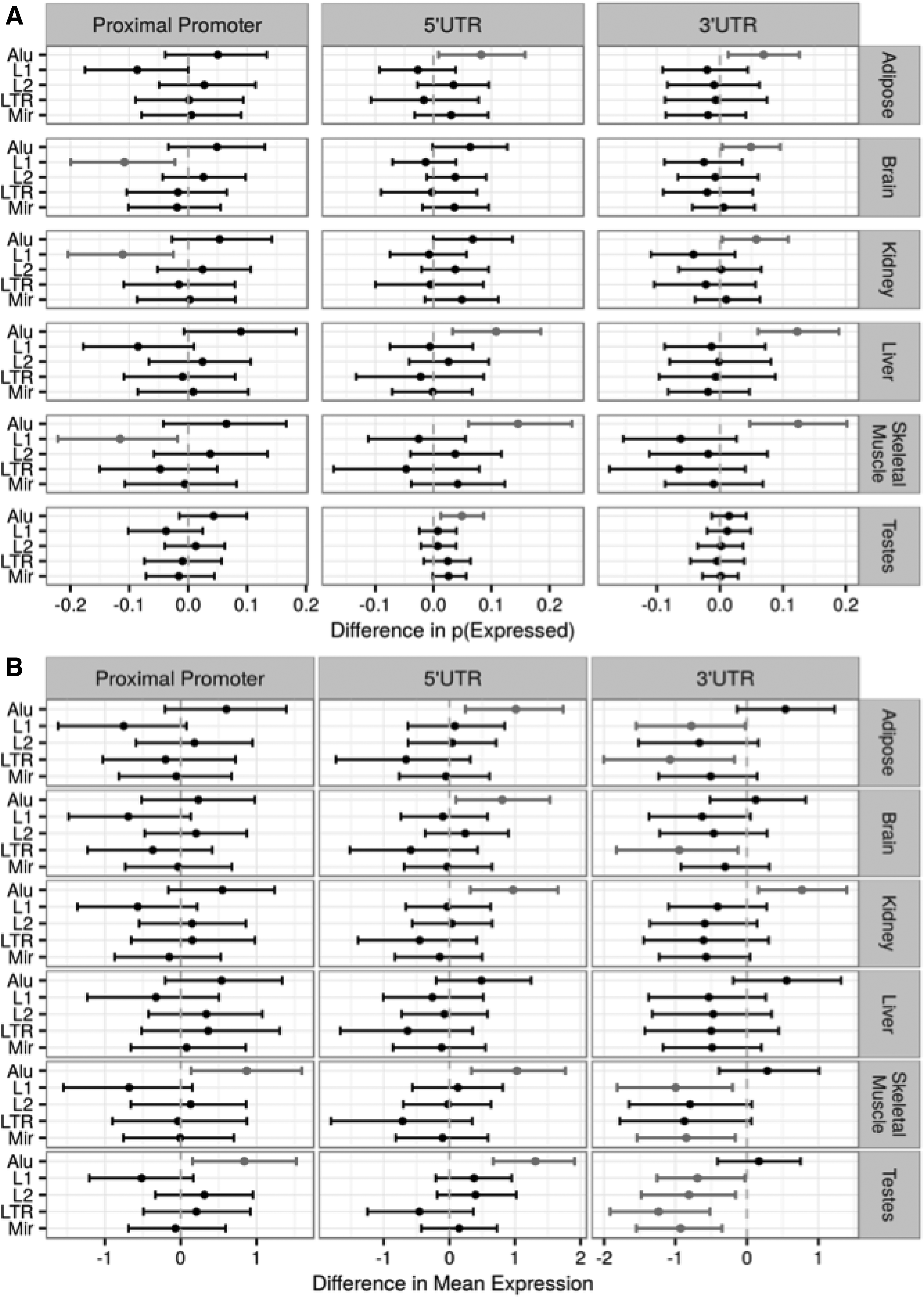
Effects on gene expression of the presence of specific TEs in each gene compartment. As TEs are far less frequent in CDS regulatory regions (Supplementary Fig. S1), the subsequent analysis focused on proximal promoters, 5′UTRs and 3′UTRs.
3.5. Effects on levels of gene expression due to the presence of a transposable element in each gene model compartment
Again, using the weighted bootstrap approach to minimize any confounding effects of co-occurring elements, the presence of an Alu in the 5′UTR was found to be associated with increased expression levels in five of the six tissues investigated (Fig. 4B). Similarly, Alu elements in the proximal promoter were associated with increased expression in two of the tissues (skeletal muscle and testis). Alu elements in 3′UTR were associated with elevated expression levels only in the kidney sample. The presence of L1, L2, and MIR elements showed varying degrees of reduced gene expression across the tissues when located in the 3′UTR only.
3.6. Analysis of genes with exapted or exonized transposable elements
To determine if exapted or exonized TEs might influence gene function, genic regions that overlapped with TEs were analyzed to assess the association of TEs with different gene functions (annotated using Gene Ontology, Supplementary Note—Section 1.5).
There are three fundamental GO categories: cellular component, molecular function, and biological process. Enrichment information for each GO category is listed in Supplementary Table S3. Analysis of the biological process (Fig. 5, and Supplementary Table S3a) shows a similar pattern with respect to the cellular component (Supplementary Fig. S4, and Supplementary Table S3b) and molecular function (Supplementary Fig. S5 and Supplementary Table S3c), repeats in 5′UTR were predominantly enriched in intracellular/cytoplasmic structures and associated with binding activities, respectively.
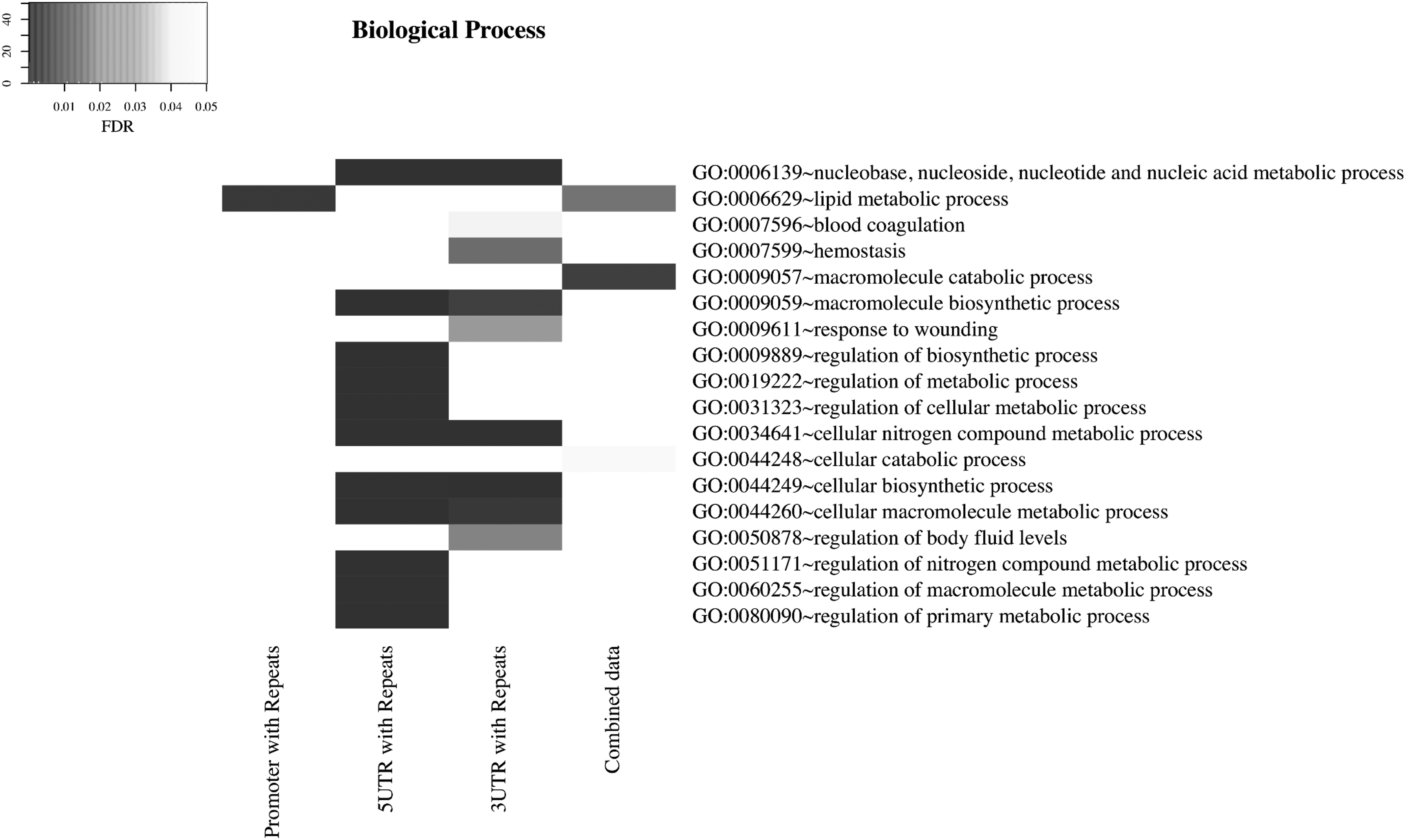
Enrichment of GO terms of genes containing TEs in promoter, 5′UTR and 3′UTR regions. Enrichment of GO terms of genes containing TEs in the biological process. Genes containing different types of repetitive elements in the proximal promoter regions are labeled as promoter with repeats, and genes containing repetitive elements in UTR regions are labeled as 5/3UTR with repeats. Genes named as combined repeats are the combined data from the three regions we mentioned above. The darker the color, the greater the GO term enrichment as determined by FDR. Figure 5 represents that 5′UTR regions containing repeats showed a strong enrichment in genes with regulatory processes, mainly involving metabolic processes, while promoter regions containing repeats were rarely associated with the biological process. GO, Gene Ontology.
The same method was applied to investigate the association of repeats with function in protein coding regions (CDS). We found that genes with CDS containing Alus were enriched for the GO term, intracellular nonmembrane-bounded organelle. Interestingly, these exonization/exaptation events were found to be associated with splice variants incorporating Alu sequences (Supplementary Table S4 and Supplementary Figs. S6 and S7).
Moreover, according to our analysis of TEs and alternative splicing data, we found that 2.98% of alternatively spliced transcripts contained TEs within protein coding exons (Table 3, Supplementary Note—Section 1.6). Alu and MIR were the two most common repeats to be involved in alternative splicing/exonization, but LINEs and LTR also contributed to a significant proportion of exonization.
Repeats were counted only if they overlapped CDS-exon regions by at least 25 bp.
AS, alternative splicing; TE, transposable element.
4. Discussion
It is known that epigenetic modification of TEs can regulate transcription by altering active chromatin, and this chromatin modification has evolved from epigenetic defense mechanisms to silence TE activity (Slotkin and Martienssen, 2007). A role for TEs in silencing is consistent with our finding that they are enriched in polycomb-repressed regions and insulators (Fig. 2). However, enrichment of TEs in active REs (Fig. 2) is suggestive of exaptation of TEs as active regulators of gene transcription. The greater proportion of p(TE|RE) in the 5′UTR and 3′UTR compared with coding exons (Fig. 2) is not surprising considering the potential adverse effect of TE insertion in a protein coding sequence (Belancio et al., 2006; Belancio et al., 2008).
TEs have been implicated in altering gene expression through a number of mechanisms (Cordaux and Batzer, 2009; Polak and Domany, 2006). However, it is difficult to analyze the impact of specific TEs on gene expression due to the co-occurrence of other TE types in or near individual genes. To overcome this problem, the bootstrapping approach was used for gene expression analysis, effectively allowing us to focus on genes containing single TE types. Figure 4B and Supplementary Figure S3B summarize our findings with regard to gene expression and indicate that Alu elements in genic regions are commonly associated with increased gene expression. Moreover, Alu elements were found to have a high probability of contributing to REs (Fig. 3). These results are consistent with previous reports that motifs in TEs such as Alus can be exapted as transcription factor binding sites and are involved in gene regulation (Kim et al., 2004; Levanon et al., 2004; Lin et al., 2009). However, in contrast to our finding that Alu elements were associated with increased gene expression, the exonization of Alu sequences from alternative splicing seems to cause only decreased expression of the alternatively spliced transcript (Lin et al., 2009).
Furthermore, when we examined the functional annotation of repeat containing genes, we found that some functions were over-represented (Supplementary Table S3). Perhaps the most interesting of these associations was that genes with Alu insertions were found to contribute to coding exons through alternative splice variants (Lewis et al., 2003). One explanation of this observation is that Alu-induced alternative transcripts may result in nonsense-mediated decay of alternative transcripts. Two examples of alternatively spliced genes of this type with implications for human disease are DISC1 and NOS3 (Supplementary Table S4 and Supplementary Figs. S6, S7). DISC1 alternative transcripts are known to contribute to increased risk of schizophrenia (Callicott et al., 2005; Pacanowski et al., 2009) and NOS3 transcript variants are associated with cardiovascular disease phenotypes. Based on the previous research, nearly 1.4% of protein coding sequences include Alu insertions (Nekrutenko and Li, 2001). Therefore, Alu exonization in protein coding genes may play an important role in modifying gene expression.
Although L1 elements are the most abundant repeats in the human genome, they were associated with decreased gene expression (Fig. 4), and were less prevalent in REs or active chromatin, when compared with other repeat classes (Fig. 3). Considering the size of L1 elements (∼6 kb), it is easy to understand that the insertion of L1 elements, even if truncated, is likely to cause disruption of host gene function (Meischl et al., 2000; Moran et al., 1999; Perepelitsa-Belancio and Deininger, 2003). Therefore, newly inserted L1 elements are probably under negative selection in regulatory regions and are less likely be exapted as an RE.
L2 and MIR are two ancient TE families conserved in mammals and are inactive or fossil TEs (Deininger and Batzer, 2002). According to previous studies, MIR elements have been exapted as functional enhancers in the human genome and are enriched in transcription factor binding sites (Jjingo et al., 2014). However, our results showed that MIR and L2 elements were associated with reduced gene expression when located in 3′UTRs (Fig. 4). This indicates that their exaptation as enhancers has the opposite effect of their exaptation within gene models. Figure 4 also showed that although endogenous retrovirus-like (ERVL) LTRs have been reported to provide molecular mechanisms for stochastic rewiring of gene expression and evolution in the mammalian germline (Franke et al., 2017), our analysis suggests that this rewiring does not affect the majority of expressed genes except for LTRs inserted into 3′UTR, which are associated with decreased gene expression.
While there are many publications implicating TEs in the regulation of individual genes, our work confirms that this role of TEs is significant across the genome. In general, most TEs would appear to be strongly associated with repression of gene expression through 5′UTR. However, the presence of Alus in 3′UTR and proximal promoter regions may act to increase gene expression. These results are supported by previous studies and provide a new understanding of how repeats are associated with epigenetic regulation of gene expression. Finally, while exapted TEs may contribute to generation of transcripts that undergo nonsense-mediated decay as part of gene regulation, we speculate that they may also provide an opportunity for alternative splicing and novel exaptation. TEs therefore are important agents of change with respect to the evolution of gene expression networks.
Footnotes
Acknowledgments
The authors wish to thank Dan Kortschak, Atma Ivancevic, Joy Raison, Reuben Buckley, and Sim Lin Lim for valuable discussions and critical reading of drafts. This work was supported by grants from the National Natural Science Foundation of China (61272250 and 61472246) and the National Basic Research Program of China (2013CB956103). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors' Contributions
D.L.A. and C.C.W. conceived, designed, and managed the study. L.Z. collected the datasets, implemented the analysis pipeline, and analyzed the data. S.M.P. analyzed the data. Z.P.Q., D.F.C., and Z.Q.H. prepared datasets. L.Z., D.L.A., and C.C.W. wrote and revised the manuscript. All authors reviewed and approved the final manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.