
Abstract
Abstract
We introduce GATTACA, a framework for fast unsupervised binning of metagenomic contigs. Similar to recent approaches, GATTACA clusters contigs based on their coverage profiles across a large cohort of metagenomic samples; however, unlike previous methods that rely on read mapping, GATTACA quickly estimates these profiles from kmer counts stored in a compact index. This approach can result in over an order of magnitude speedup, while matching the accuracy of earlier methods on synthetic and real data benchmarks. It also provides a way to index metagenomic samples (e.g., from public repositories such as the Human Microbiome Project) offline once and reuse them across experiments; furthermore, the small size of the sample indices allows them to be easily transferred and stored. Leveraging the MinHash technique, GATTACA also provides an efficient way to identify publicly available metagenomic data that can be incorporated into the set of reference metagenomes to further improve binning accuracy. Thus, enabling easy indexing and reuse of publicly available metagenomic data sets, GATTACA makes accurate metagenomic analyses accessible to a much wider range of researchers.
1. Introduction
D
A common first step in such pipelines is de novo assembly of metagenomic reads into contigs. Since this initial assembly typically does not produce fully reconstructed microbial genomes, the partially assembled contigs are then deconvoluted using metagenomic binning, which groups together contigs that belong to the same microorganism. Current binning techniques fall into mainly two categories: (1) supervised classification of contigs into known taxons via comparisons to previously cataloged species (Huson et al., 2007; Rosen et al., 2011; Wood and Salzberg, 2014) and (2) unsupervised clustering techniques using features derived directly from the metagenomic sample data (Kislyuk et al., 2009; Albertsen et al., 2013; Alneberg et al., 2014; Wu et al., 2014; Kang et al., 2015; Lu et al., 2016; Luo et al., 2016), where unsupervised clustering has the clear advantage of binning contigs that pertain to previously unknown species. While some unsupervised techniques (Kislyuk et al., 2009; Strous et al., 2012) perform clustering based only on the contig sequence composition (the frequency of certain short motifs, e.g., all tetramers), the most successful recent approaches (Albertsen et al., 2013; Alneberg et al., 2014; Wu et al., 2014; Kang et al., 2015; Lu et al., 2016) also incorporate contig coverage profiles across multiple metagenomic samples.
In brief, given a cohort of metagenomic samples, these techniques compute the average coverage of each contig in each sample by mapping sample reads to the contigs using standard read mapping tools, such as Bowtie2 (Langmead and Salzberg, 2012). Naturally, contigs belonging to the same species will have similar abundances across different samples, resulting in similar coverage profiles, which allow them to be clustered together. This approach is accurate but has two main limitations: it requires a large cohort of samples, as well as sizable compute resources for read alignment. We address both of these limitations in this work.
In particular, we present GATTACA, a lightweight framework for metagenomic binning, which (1) avoids read alignment without loss of accuracy and (2) enables efficient stand-alone analysis of single metagenomic samples. Both results are based on the finding that we can approximate contig coverages using kmer counts while still achieving the same binning accuracy as leading alignment-based methods. In addition to offering a significant speedup in coverage estimation, using kmer counts, as opposed to alignment, provides us with the exciting ability to index offline any publicly available metagenomic sample and incorporate it into the coverage profile of the contigs being processed. This allows us to efficiently pull in data from large growing repositories, such as the Human Microbiome Project (HMP; Turnbaugh et al., 2007) or EBI Metagenomics archive (Hunter et al., 2014), into any metagenomic study (especially one where only a single or few samples are available) at almost no cost. For example, the kmer count index of an HMP sample used in our experiments (containing 20 million reads) only requires: 150 MB on average. We achieve the small space requirement by leveraging memory-efficient hashing with minimal perfect hash functions (MPHFs) and the probabilistic Bloom filter data structure. In contrast, using these data sets with read alignment would require much larger downloads and expensive subsequent handling to map the reads.
While using small indices allows us to incorporate a large number of publicly available samples into a given study, not all existing samples will carry content relevant to the study in question. Namely, samples that do not contain any of the species present in a given set of contigs cannot contribute any useful information for grouping the contigs. The same logic applies also to samples that carry content identical to a sample that has already been included. This motivates the need to additionally define appropriate sample selection criteria, for which we propose two metrics: (1) relevance and (2) diversity. More specifically, we would like to select a panel of samples that share content with the samples being analyzed (our query) but that also differ in the content that is shared. We use the MinHash technique (Broder et al., 2000) to find such samples efficiently. At a high level, we create and index small MinHash fingerprints for each sample in the database (offline) and then extract the appropriate samples according to the fingerprint of the query. The resulting index can be separately downloaded and used to determine which samples to include into the analysis; it needs to be updated only occasionally when new samples become available.
We evaluate GATTACA in clustering contigs assembled across multiple samples (coassemblies) and from individual samples, using both synthetic and real data sets. We compare our results with several state-of-the-art methods in metagenomic binning: CONCOCT (Alneberg et al., 2014), MetaBAT (Kang et al., 2015), and MaxBin (Wu et al., 2014), using standardized cluster evaluation metrics and benchmarks (reusing evaluation scripts from existing methods when appropriate). GATTACA was implemented in C++ and Python and is freely available at https://github.com/viq854/gattaca-public.
2. Methods
2.1. Index of kmer counts
To quickly estimate contig coverages, GATTACA builds a small index of kmer counts for each sample in the cohort. Many solutions have been proposed for exact [e.g., using hash tables (Marçais and Kingsford, 2011; Rizk et al., 2013) or minimum perfect hash functions (Patro et al., 2014)] and approximate kmer counting [e.g., using the count-min sketch (Zhang et al., 2014)]. Since the content of each sample in our panel is static, our index uses an MPHF (Cichelli, 1980) to store the kmer counts without loss of accuracy, resulting in a drastic reduction in space when compared to traditional hash tables (we also found it to be more space efficient than the count-min sketch solution for the same binning accuracy). At a high level, given a set S of n keys, an MPHF h provides a mapping between the keys and n consecutive integers from 0 to
2.1.1. Index construction
To construct the index, we first generate the kmers from all the reads in the sample (accounting for both forward and reverse complement strands) and exclude kmers that occur only once, since these are most likely present due to sequencing errors. We use a kmer length of 31-bp in our experiments (compacting the kmers into 64-bit integers for convenience). We then generate the MPHF, hS, for the resulting set of distinct kmers, S, and store their counts in an integer array A
Finally, we need to store the elements in S to support lookups, since
2.1.2. Coverage estimation
Given a contig c and an index I of a cohort sample, we estimate the coverage of c in this sample by performing lookups in I for each kmer in c and then computing the median of the resulting counts. More specifically, we return the median of the set of counts
2.2. Contig representation
Given a set of contigs assembled from a single or multiple metagenomic samples, our goal is to bin together the contigs that belong to the same class (e.g., species or strain). Similar to existing methods, for example, CONCOCT, we first represent each contig as a multidimensional vector using both its sequence composition and coverage profile across multiple samples, where our coverages are approximated using kmer counts instead of read mapping, as previously described. Namely, given M reference samples (either from the same study or from a public database), our coverage profile is the median count of the contig kmers in each sample, while the composition profile is the normalized frequency of each possible tetramer in the contig and its reverse complement (resulting in a total of
2.3. Clustering algorithm
Given the resulting vector representations, we cluster the contigs using a Bayesian Gaussian mixture model (GMM) with a Dirichlet prior. In brief, we define a mixture distribution p of K Gaussian components over n data points
and a prior term
Here,
We perform inference by maximizing the marginal log-likelihood
over the set of approximating distributions q. By our choice of conjugate prior, the posterior
At a high level, the above model is very similar to automatic relevance determination (ARD), which is used by CONCOCT. We have found this approach to perform slightly better in practice than ARD, especially for automatically determining the number of clusters in the data; however, both algorithms are implemented in our software package and can be experimented with. Other alternative clustering methods can also be easily plugged into GATTACA's binning pipeline.
2.4. Sample selection
Given a query sample, Q, we would like to select samples from the public database, which can provide discriminatory features for clustering the contigs of Q (where the features represent the coverage of the contigs in the respective samples). Intuitively, the selected samples must share some content with Q (have relevance), as well as have pairwise diversity among themselves to guarantee coverage of different contigs of Q. Similar relevance and diversity concepts can be found in online recommendation systems [e.g., for articles or music (Abbar et al., 2013; Chen and Cong, 2015)].
A standard approach for comparing strings is to represent them as sets and then measure the relative size of their sets' intersection. For example, text documents can be represented as sets of words, while DNA sequences are commonly represented as sets of overlapping kmers. In this case, the similarity of two given sets A and B is measured using the Jaccard coefficient
Due to its simplicity and efficiency, it has also been used for various tasks involving DNA sequence comparisons, such as motif discovery (Wijaya et al., 2005), de novo assembly (Berlin et al., 2014), read alignment (Popic and Batzoglou, 2017), and clustering of DNA sequence collections, including metagenomic sequences (Ondov et al., 2016). Here we use it to quickly estimate the relevance and diversity of the set of metagenomic samples considered for inclusion into the binning panel. Internally, GATTACA uses the implementation of MinHash for DNA sequence comparisons adapted from Popic and Batzoglou (2017). At a high level, we first breakdown the reads of a sample Q into a set of overlapping kmers,
We define the relevance score between a sample A and B as simply the ratio, r, of the number of entries shared by their fingerprints over L (which approximates their original Jaccard similarity; Broder et al., 2000). Then, the user can include all samples S for which
Finally, if the number of relevant samples is too high, the panel can be reduced using the diversity criterion. That is, given all the relevant samples, we can select n samples that maximize the diversity of the set. This problem is known as the dispersion problem (Erkut, 1990), where the objective is to locate k points among n, such that some function of distances between the k points is maximized. One popular optimality criterion is the MAX–MIN, which maximizes the minimum distance between a pair of points. This problem is known to be NP-hard; however, an efficient greedy heuristic algorithm exists for the MAX–MIN dispersion problem when the distances satisfy the triangle inequality, with provable performance guarantee of 2 (Ravi et al., 1991). Given two samples A and B, we define their diversity as
3. Results
3.1. Data sets
3.1.1. Synthetic data sets
We used two synthetic data sets generated by Alneberg et al. (2014) from the 16S rRNA samples of the HMP (Turnbaugh et al., 2007). The first data set (“Species-Mock”) consists of 96 samples containing a mixture of 101 different species (without strain-level variation), while the second data set (“Strain-Mock”) consists of 64 samples comprising a mixture of 20 different organisms, of which some represent strains of the same species (e.g., this data set includes five different E. coli strains). The relative abundance profiles of the species and strains in each sample were assigned according to the distribution of the 101 and 20 most abundant organisms in the original HMP samples, respectively. Reads (100-bp long) were simulated from random positions of the genomes present in the sample based on their relative abundance, for a total of 7.75 million reads and 11.75 million reads in each “Species-Mock” and “Strain-Mock” sample, respectively. Both data sets contain the set of contigs coassembled across all the samples by Alneberg et al. (2014) using the Ray assembler (Boisvert et al., 2010), and partitioned into fragments of 10 kb when appropriate.
We used the default minimum contig length of 1000-bp when running CONCOCT, MaxBin, and GATTACA; this parameter was set to 1500-bp for MetaBAT, which is the smallest length supported by this method. As a result, the “Species-Mock” included 37,627 valid contigs and the “Strain-Mock” included 9411 valid contigs (for all tools except MetaBAT). To determine the genome assignment for each contig, the simulated reads were mapped back to the contigs and the genome was selected based on the majority of the mapped reads (with the requirement that it corresponds to at least 50% of the reads and there are at least 10 unambiguously mapped reads); this is the same criteria used by Alneberg et al. (2014).
To evaluate single-sample binning, we also assembled the contigs for several simulated samples of the “Species-Mock” independently. We used the SPADES (Bankevich et al., 2012) assembler with default parameters and automatic kmer selection, and applied the same postprocessing and labeling criteria.
3.1.2. Real data sets
To evaluate coassembly-based binning, we used the small Sharon et al. (2013) data set comprising 11 fecal samples from a preterm infant (Sharon et al., 2013). We downloaded 1.372 × 108 100-bp short reads from the SRA052203 NCBI archive as 18 separate samples (of which 7 were resequenced from the original 11 samples). The coassembled contigs for this data set were made public by Alneberg et al. (2014). To test binning accuracy, we used the CheckM (Parks et al., 2015) method based on single-copy core genes (SCGs), which are housekeeping genes that occur in single copies in the microbial genomes and have been previously used to evaluate the purity of the metagenomic clusters (Alneberg et al., 2014).
To evaluate binning of contigs assembled from single samples, we downloaded Illumina raw sequencing reads from the following two publicly available gut microbiome sample collections: (1) gut samples from HMP (148 total) and (2) 56 gut samples from the study of Albertsen et al. (2013) available through the European Nucleotide Archive. We used the SPADES assembler (Bankevich et al., 2012) with default parameters to assemble the contigs of individual samples (cutting contigs into 10 kb fragments and filtering contigs shorter than 1000-bp, as in simulation). Since the HMP samples were very large (over 30 GB/sample uncompressed), we subsampled them using the seqtk program to 20 million paired-end reads per sample during index construction. To label the contigs in each evaluated sample, we used the TAXAssign program. Labels were assigned based on the top 100 hits against the BLAST NCBI genome database that had at least 95% identity and 90% query coverage. A taxonomic label was assigned if 90% of the hits at a given level belong to the taxon associated with that label. This procedure classified roughly 15% of them on average at the species level depending on the sample. Since only a small fraction of contigs could be classified via TAXAssign, we also used SCGs to evaluate binning accuracy with CheckM.
3.2. Clustering evaluation metrics
We apply the following three standardized metrics for evaluating metagenomic clustering of labeled contigs: (1) recall—measures how many same-class contigs are placed in the same cluster, (2) precision—measures the purity of each cluster, and (3) adjusted Rand index (ARI)—measures how often pairs of same-class contigs are clustered together. On synthetic data sets (for which we know the ground-truth genome assignment) and the contigs confidently classified using TAXAssign, we compute these metrics using the Validate.pl script provided by CONCOCT directly (Alneberg et al., 2014). For SCG-based evaluation, we report the metrics computed by CheckM, using the “benchmark.R” script provided by MetaBAT (Kang et al., 2015) for visualization.
3.3. Synthetic benchmarks
First, we show that replacing read mapping with kmer counting for coverage estimation does not result in loss of clustering accuracy, using two synthetic benchmarks created by Alneberg et al. (2014). Namely, we compare the performance of GATTACA, CONCOCT, MaxBin, and MetaBAT on the “Species-Mock” and “Strain-Mock” data sets, using the published coassembled contigs. Here, we built the index of kmer counts only from the cohort of samples associated with each data set. As a result, our “Species-Mock” index contains 96 samples (with a total size of 12 GB) and our “Strain-Mock” index contains 64 samples (size of 5.1 GB). Table 1 shows the accuracy results for each tool. GATTACA achieves similar accuracy to CONCOCT and MaxBin (all higher than MetaBAT) on both data sets, with a somewhat lower recall and higher precision and ARI. MetaBAT achieves the highest precision on both data sets; however, it clusters substantially fewer contigs.
Evaluation metrics were computed with the Validate.pl script provided by CONCOCT.
ARI, adjusted Rand index.
Since one of the goals of this project is to support single-sample analysis, we also evaluated single-sample contig clustering on simulated data. In particular, we assembled several samples of the synthetic “Species-Mock” data set separately (which resulted in 11 thousand contigs on average per sample) and used the remaining samples to build the index of kmer counts in a “leave-out-out” manner; namely, for a given sample Q, the index was built from all the samples in the original cohort except Q. Table 2 shows GATTACA's performance on three different samples. As expected, the accuracy results are somewhat lower but still comparable to the coassembly benchmark.
Contigs were assembled from individual samples of the “Species-Mock” data set and clustered using a reference index built from all other samples in this data set.
3.4. Real data benchmarks
Next, we evaluate the binning performance of each method on the coassembly of the real Sharon et al. (2013) data set. Since the true reference genomes are not available for this experiment, we used the CheckM (Parks et al., 2015) method based on SCGs to approximate recall and precision. The GATTACA index was built from the 18 samples of the data set, having a total size of 2.2 GB. Figure 1 shows the number of genomes identified by each method with a precision higher that 90% and 95%, respectively, and varying recall, as reported by CheckM. All methods, except MetaBAT, produce three highly accurate genomes with precision and recall ≥95% (MetaBAT generates only one such genome). GATTACA's performance is very similar to CONCOCT here, with the exception of an unreported bin at precision ≥90%, which CONCOCT produces with a recall of 30%. Figure 2 shows how these performance metrics vary across the generated bins.

Binning performance on the real Sharon data set reported by CheckM. The number of genomes identified with ≥90% (left) and ≥95% (right) is shown for varying recall. The image was generated using the “benchmark.R” script (slightly modified) provided by MetaBAT.
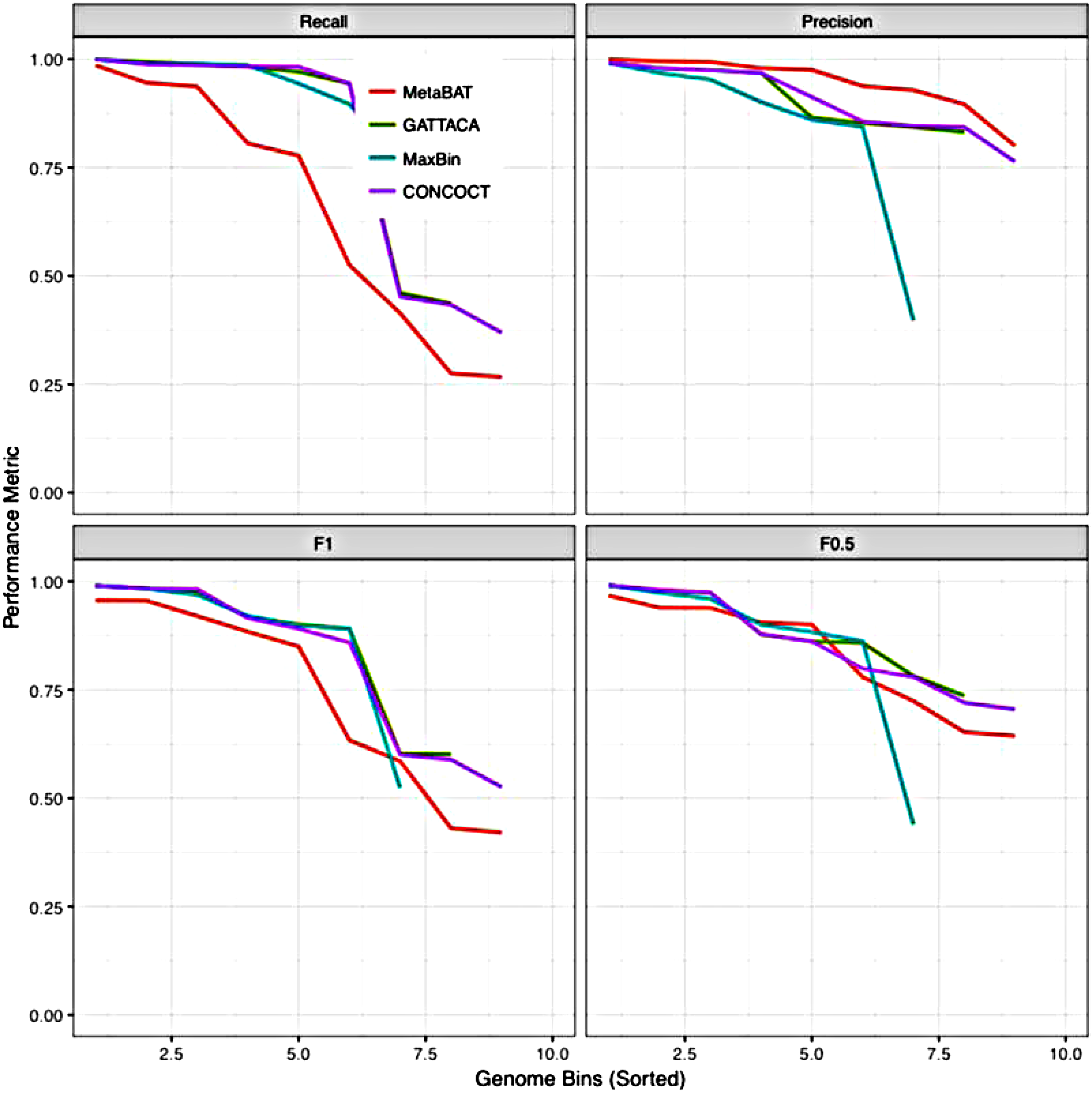
Binning performance on the real Sharon data set reported by CheckM. The image was generated using the “benchmark.R” script (slightly modified) provided by MetaBAT.
We also evaluate the binning of single-sample assemblies using a panel of publicly available data sets of the gut microbiome. Table 3 shows the binning accuracy computed using TAXAssign labels for three HMP stool samples clustered using a panel of 95 different stool samples from the HMP database. Due to the long read mapping runtime, we show results for CONCOCT, MetaBAT, and MaxBin on only one of the HMP samples, SRS011239. GATTACA's performance is overall stable and accurate across samples, with an ARI of about 90%. On SRS011239, GATTACA achieves an accuracy similar to that of other methods. Figure 3 also shows the SCG-based results for SRS011239 obtained using CheckM.
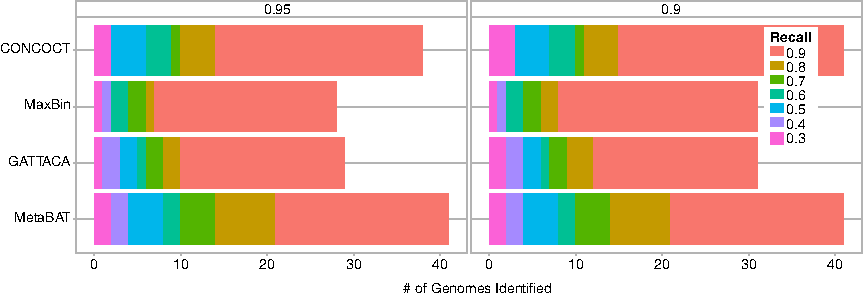
Binning performance on the real SRS011239 sample and a panel of 95 HMP metagenomes reported by CheckM. The number of genomes identified with ≥90% (left) and ≥95% (right) is shown for varying recall. The image was generated using the “benchmark.R” script (slightly modified) provided by MetaBAT. HMP, Human Microbiome Project.
Contigs that were not covered by at least a single sample were not included in the clustering step.
Finally, we evaluate GATTACA in clustering contigs from a single sample of the Albertsen et al. (2013) study using an index built from 46 samples of the study and an index augmented with 83 HMP samples selected using the relevance criteria previously described. Table 4 shows the results based on the TAXAssign labels. As previously shown by Alneberg et al. (2014), binning accuracy increases with larger cohort sizes. Similarly, in this experiment, it can be seen that the binning precision improves with the addition of the HMP samples, motivating the inclusion of publicly available data in studies with fewer available samples.
Contigs that were not covered by at least a single sample were not included in the clustering step.
HMP, Human Microbiome Project.
3.5. Runtime and space consumption
To estimate the reduction in runtime and space consumption of our approach, we compare the runtime and space requirements of the CONCOCT pipeline, which is based on read mapping [internally using Bowtie2 (Langmead and Salzberg, 2012), samtools (Li et al., 2009), and Picard tools], to the GATTACA kmer counting implementation for computing the coverage of preassembled contigs. The reported runtimes were obtained on a single 2.67 GHz Intel Xeon X5550 processor, in single-threaded mode, although both of the tools can be easily run in parallel. We outline the performance across several data sets hereunder.
For the “Species-Mock” data set, coverage estimation in one given sample with the CONCOCT pipeline took 62 minutes; with an additional 16 minutes for the initial sample download from the repository. This single sample also requires 3 GB (uncompressed)/580 MB (compressed) disk space to be stored in FASTA format (without base qualities), with an additional 450 MB to store its alignment results. With GATTACA, the kmer count lookups in this sample took 2.7 minutes and the GAC index construction took 6 minutes. Ignoring the download time, the resulting speedup in coverage estimation is 23 × if the index for this sample has already been precomputed offline; otherwise, the speedup is 7 × when the index is computed on the fly. Moreover, the GAC index only takes 140 MB and it is the only data structure required to use this sample for binning.
For the “Strain-Mock” data set, coverage estimation in one given sample with the CONCOCT pipeline took 101 minutes, with an additional 24 minutes for the initial sample download from the repository. This single sample also requires 4.8 GB (uncompressed)/842 MB (compressed) disk space to be stored; with an additional 685 MB to store its alignment results. With GATTACA, the kmer count lookups in this sample took 0.75 minutes and the GAC index construction took 6.2 minutes. Therefore, the resulting speedup in coverage estimation is 135 × with offline indexing and 14.5 × speedup with online indexing. Moreover, the GAC index only takes 91 MB.
Finally, for the real HMP data set, coverage estimation in one given sample (containing 20 million reads) with the CONCOCT pipeline took 51.2 minutes, while GATTACA took 2.7 minutes for lookups and 5.3 minutes in GAC index construction, resulting in a 19 × speedup with offline indexing and 6.4 × speedup with online indexing. This sample also requires 7 GB (uncompressed)/2.6 GB (compressed) of disk space [with an initial download size of 7 GB (compressed) before subsampling]. On the contrary, its resulting GAC index only requires 108 MB.
Given these results and considering that binning panels usually consist of one hundred or more samples, both the runtime and disk savings for switching to GATTACA can be very substantial. Note, the reported performance was obtained using the BDZ algorithm (Botelho et al., 2007) for index construction, and our recent preliminary results show that using the BBhash algorithm (Limasset et al., 2017) can speed up the runtime further.
3.6. Evaluation of sample selection
We performed the following experiment to evaluate the performance of MinHash for sample selection. Given a randomly selected real query sample Q (namely, SRS011134 from the HMP), we created a panel of 50 samples (each containing 10 M reads of length 100-bp) that share a varying fraction of reads with Q, ranging from 2% to 100%. More specifically, we first started with Q and 49 samples containing reads simulated from the chicken genome (under the assumption that there will be little content similarity between this genome and the species present in SRS011134). We then replaced varying fractions of the chicken reads with the reads of SRS011134. Figure 4 shows the true Jaccard similarity between the panel samples and Q, as well as the Jaccard similarity estimated using MinHash, with varying fingerprint lengths. We found that MinHash fingerprints of length 1024 provide a good trade-off between runtime and sensitivity (fingerprint length is a configurable parameter in our software). This fingerprint length requires only 8K additional space per metagenomic sample. The fingerprint comparisons performed in this experiment took less than a second in total, and can easily scale to large publicly available metagenomic data sets. More experiments of interest on MinHash can be found in the recent article (Ondov et al., 2016); for example, it shows the results of applying MinHash to cluster HMP samples.
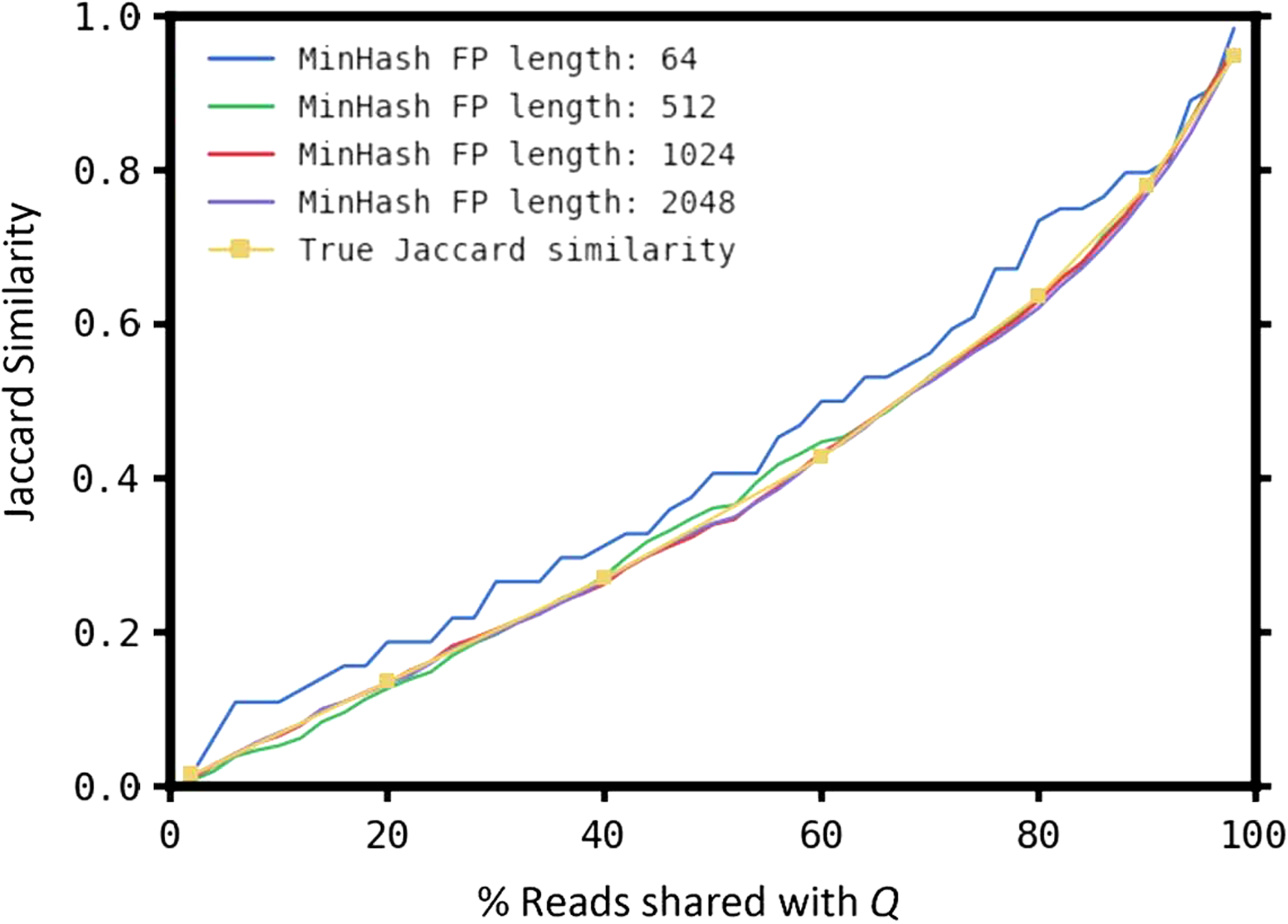
Jaccard similarities estimated using MinHash for a query sample Q (SRS011134) and a panel of 49 samples sharing varying levels of content similarity with Q. The true Jaccard similarity is shown along with estimates computed from fingerprints of different lengths.
4. Conclusion
GATTACA is a lightweight method for metagenomic binning, which achieves comparable binning accuracy with leading alignment-based methods at a fraction of the cost by approximating contig abundances using kmer counts. Furthermore, through offline indexing of publicly available cohorts of metagenomes, it enables efficient analysis of single metagenomic samples, catering to settings with scarce computational and data resources. As a result, an ongoing effort of this project is to index all the publicly available samples from various metagenomic archives and make them available to the public.
Footnotes
Acknowledgments
V.K. was supported by a VMWare Stanford Graduate Fellowship. The authors thank Roye Rozov, Rayan Chikhi, Johannes Alneberg, and the anonymous reviewers for their helpful feedback on the article and/or the software.
Author Disclosure Statement
S.B. is a cofounder and advisor to DNAnexus and member of the scientific advisory boards of 23andMe, Eve Biomedical, and GenapSys. S.B. and V.P. are currently employed by Illumina, Inc. M.S. is a cofounder of Personalis and a member of the scientific advisory boards of Personalis, AxioMx, and GenapSys.