2′-O-methylation plays an important biological role in gene expression. Owing to the explosive increase in genomic sequencing data, it is necessary to develop a method for quickly and efficiently identifying whether a sequence contains the 2′-O-methylation site. As an additional method to the experimental technique, a computational method may help to identify 2′-O-methylation sites. In this study, based on the experimental 2′-O-methylation data of Homo sapiens, we proposed a support vector machine-based model to predict 2′-O-methylation sites in H. sapiens. In this model, the RNA sequences were encoded with the optimal features obtained from feature selection. In the fivefold cross-validation test, the accuracy reached 97.95%.
1. Introduction
2′-O-methylation is catalyzed by the 2′-O-methylation transferase. In the reaction, a methylation group is added to the 2′-hydroxyl group of the ribose moiety of a nucleotide (Fig. 1; Kiss, 2002). 2′-O-methylation is one kind of post-transcriptional modification in various cellular RNAs and plays critical roles in the regulation of gene expressions at the post-transcriptional levels (Bachellerie et al., 2002).
Schematic diagram of 2′-O-methylation.
When the 2′-O-methylation sites accumulate around the functional region of ribosomal RNA (rRNA), they affect the ribosome structure and function (Decatur and Fournier, 2002). When the ribose 2′-O-methylation occurs in the cap structure of the messenger RNA (mRNA), the RNA sensor Mda5 (Zust et al., 2011) can distinguish between itself and nonautologous mRNA. In addition, the 2′-O-methylation at the 3′ end of Piwi-interacting RNA (piRNA), endo-small interfering RNAs, and microRNAs can protect the molecules from uridine (Li et al., 2005) and exonuclease degradation and regulate specific RNAi pathways (Ramachandran and Chen, 2008).
Although 2′-O-methylation had been explored, the mechanism of 2′-O-methylation in mRNA is still unclear (Maden, 2001). Therefore, it is necessary to reveal the mechanism of RNA 2′-O-methylation.
Accurate identification of 2′-O-methylation sites is the key step in understanding its regulatory mechanism. Many biochemical approaches have been developed to detect and analyze RNA 2′-O-methylation (Dong et al., 2012), such as liquid chromatography coupled with mass spectrometry and two-dimensional thin-layer chromatography approaches. Recently, the RTL-P method based on reverse transcription at low dNTP concentrations and PCR was proposed to identify both previously characterized and novel 2′-O-methylated sites in human rRNA, yeast rRNA, and mouse piRNAs.
Although many details of the 2′-O-methylation sites can be obtained in biochemical experiments, the experimental techniques and processes are often time consuming and expensive. With the large number of biological sequences produced in the postgenome era, it is necessary to develop the computational method to identify 2′-O-methylation sites (Zust et al., 2011; Sun et al., 2016). Thus, the machine-based approach is usually quite robust and effective in dealing with various biological problems and becomes a complementary method to the experimental technique.
In this study, based on the experimental 2′-O-methylation data of Homo sapiens, we presented a support vector machine (SVM)-based model to predict 2′-O-methylation sites in H. sapiens. The minimal redundancy maximal relevance (mRMR) was used to pick out the over-represented features. In the fivefold cross-validation, our method achieved an overall accuracy of 97.95%.
For the convenience of the scientific community, a freely accessible web server for the proposed method is provided at http://lin-group.cn/server/iRNA-2OM.
2. Methods
2.1. Benchmark data set
The original data containing experimentally validated 2′-O-methylation sites in H. sapiens were downloaded from RMBase (Sun et al., 2016). The RMBase (http://mirlab.sysu.edu.cn/rmbase) is a database of RNA-modified genome-wide data identified in 18 independent studies of high-throughput modified data.
In the previous study (Chen et al., 2016b), based on RMBase, to avoid redundancy and reduce homology deviations, 80% similarity RNA sequences were removed by using the CD-HIT procedure (Fu et al., 2012). Finally, a total of 147 2′-O-methylation site contained sequences in H. sapiens were obtained and treated as positive samples. These sequences are all 41-nt long with the 2′-O-methylation site in the center.
The negative samples were produced by selecting the 41-nt long sequences, in which the central nucleotides were not 2′-O-methylated. A large number of negative samples could be obtained. It is well known that an imbalanced data set can significantly affect the evaluation results of the proposed models. Therefore, 147 negative samples were randomly picked out to balance positive and negative samples. Thus, the final benchmark data set was formulated as
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{ \rm{S}} = {S^ + }{ \rm{ \;}} \cup { \rm{ \;}}{S^ - } , \tag{1}
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \rm{S}}$$
\end{document} is the benchmark data set, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${S^ + }$$
\end{document} is the positive subset containing 147 true 2′-Omethylation site contained sequences, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${S^ - }$$
\end{document} is the negative subset containing 147 false 2′-O-methylation site contained sequences of H. sapiens, which are available at (http://lin-group.cn/server/iRNA-2OM).
2.2. Support vector machine
SVM is a supervised learning model for pattern recognition, classification, and regression analysis. It has been successfully applied in the field of bioinformatics (Cao et al., 2014; Chen et al., 2016d; Tang et al., 2016; Yang et al., 2016; Zhao et al., 2016, 2017; Zou et al., 2016a; Dao et al., 2017; Lai et al., 2017; Lin et al., 2017; Manavalan and Lee, 2017; Manavalan et al., 2017, 2018a, 2018b; Feng et al., 2018). The basic idea of SVM is to transform the data into a high-dimensional feature space and then determine the optimal separating hyperplane. Gaussian radial basis function (RBF) is a widely used kernel function due to its wonderful performance in nonline classification. Thus, the RBF kernel function was used in the study. Some software packages have been developed for reducing the programming burden of researchers, including LIBSVM, mySVM, and SVMLight (Sch and Burges, 1999; Chang et al., 2000). In this study, we used the free software the LIBSVM 3.20 package to implement SVM (Chang et al., 2000). In the SVM operation engine, the grid search method was applied to optimize the regularization parameter C and kernel parameter γ:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\left\{ { \begin{matrix} {{2^{ - 5}} \le C \le {2^{15}} \; \; \; \; \; \; \; \;{ \rm{with \;step \;}} \Delta C = 2 \; \; \; \;} \\ {{2^{ - 15}} \le \gamma \le {2^{ - 5}} \; \; \; \; \; \;{ \rm{with \;step}} \; \Delta \gamma = {2^{ - 1}}} \hfill\\ \end{matrix} } \right. \tag{2}
\end{align*}
\end{document}
2.3. Chemical properties
A RNA chain is composed of four types of nucleotides: adenine (A), guanine (G), cytosine (C), and uracil (U).
In this article, RNA chemical properties were used to encode RNA sequence (Chen et al., 2016a,b,c). According to their chemical properties, the four nucleotides can be classified into three different groups (Chen et al., 2017), as shown in Table 1.
Classification of Nucleotides
Chemical properties
Attribute
Nucleotides
Ring structure
Two rings
A, G
One ring
C, U
Hydrogen bond
Strong
A, C
Weak
G, U
Chemical functionality
Amino group
A, U
Keto group
U, G
In terms of ring structure, adenine and guanine have two rings, whereas cytosine and uracil have only one ring. When forming secondary structures, guanine and cytosine have strong hydrogen bonds, whereas adenine and uracil have weak hydrogen bonds. In terms of chemical functionality, adenine and cytosine can be classified into the same group, called amino group, whereas guanine and uracil can be classified into the keto group.
To reflect these chemical properties, we denote the i-th nucleotide as
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{N_i} = \left( {{x_i} , {y_i} , {z_i}} \right) \;. \tag{3}
\end{align*}
\end{document}
Three coordinates \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( {{x_i} , {y_i} , {z_i}} \right) { \rm{ \;}}$$
\end{document} were used to represent the three chemical properties and the value of 1 or 0 was assigned to the coordinates. Each nucleotide can be encoded by the following formula:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{x_i} = \left\{ { \begin{matrix} {1 \; \; \; \;if \;{S_i} \epsilon \left\{ {A , G} \right\} } \\ {0 \; \; \; \;if \;{S_i} \epsilon \left\{ {C , U} \right\} } \\ \end{matrix} } \right. \tag{4}
\end{align*}
\end{document}\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{y_i} = \left\{ { \begin{matrix} {1 \; \; \; \;if \;{S_i} \epsilon \left\{ {A , C} \right\} } \\ {0 \; \; \; \;if \;{S_i} \epsilon \left\{ {G , U} \right\} } \\ \end{matrix} \;} \right. \tag{5}
\end{align*}
\end{document}\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{z_i} = \left\{ { \begin{matrix} {1 \; \; \; \;if \;{S_i} \epsilon \left\{ {A , U} \right\} } \\ {0 \; \; \; \;if \;{S_i} \epsilon \left\{ {C , G} \right\} } \\ \end{matrix} } \right.{ \rm{ \;}}. \tag{6}
\end{align*}
\end{document}
Thus, A, C, G, and U can be, respectively, represented with the coordinates (1, 1, 1), (0, 1, 0), (1, 0, 0), and (0, 0, 1).
2.4. Nucleotide composition
The density di of any nucleotide nj at position i in an RNA sequence from the nucleotide composition surrounding the 2′-Omethylation site in the training data set of the sequence is defined as (Chen et al., 2016b; Feng et al., 2017):
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{d_i} = {1 \over { \left\vert {{N_i}} \right\vert }} \mathop \sum \limits_{j = 1}^l f \left( {{n_j}} \right) , { \rm{ \; \; \; \; \; \; \; \; \;}}f \left( {{n_j}} \right) = \left\{ { \begin{matrix} {1 \; \; \; \; \;if \;{n_j} = q \;} \\ {0 \; \;othercases} \\ \end{matrix} } \right.{ \rm{ \;}} \tag{7}
\end{align*}
\end{document}
where l is the sequence length, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left\vert {{N_i}} \right\vert$$
\end{document} is the length of the i-th prefix string \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left\{ {{n_1} , {n_2} , \ldots , {n_i}} \right\} $$
\end{document} in the sequence, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${ \rm{q}} \in \left\{ {A , C , G , U} \right\} $$
\end{document}.
2.5. Type 2 PseKNC
To consider the global information and the local information of the sequence, type 2 PseKNC method was adopted (Chen et al., 2014). The new feature data reflect both the local and the global sequence information of the nucleotide sequence.
The type 2 PseKNC can be expressed as
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
D = { \left[ {{d_1} , {d_2} , \cdots {d_{{4^k}}} , {d_{{4^k} + 1}} , {d_{{4^k} + 2}} , \cdots {d_{{4^k} + \Lambda }} , \cdots {d_{{4^k} + \Lambda \lambda - 1}} , {d_{{4^k} + \Lambda \lambda }}} \right] ^T} , \tag{8}
\end{align*}
\end{document}
where ω is weight factor for sequence order effect and fu is the normalized frequency expressed in Equation(10):
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{ \tau _1 } &= \frac { 1 } { { L - K - 1 } } \sum \nolimits_ { i
= 1 } ^ { L - K - 1 } \, \,H_ { i , i + 1 } ^1 \\ { \tau _2 } &=
\frac { 1 } { { L - K - 1 } } \sum \nolimits_ { i = 1 } ^ { L - K
- 1 } \, \,H_ { i , i + 1 } ^2 \\ & \, \, \, \, \, \, \, \, \, \,
\, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \,
\, \, \, \, \, \, \vdots \\ { \tau _ \Lambda } &= \frac { 1 } { {
L - K - 1 } } \sum \nolimits_ { i = 1 } ^ { L - K - 1 } \, \,H_ {
i , i + 1 } ^ \Lambda \, \, \, \, \, \, \, \, \, \, \lambda < ( L
- K ) \\ & \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \,
\, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \, \vdots
\\ { \tau _ { \Lambda \lambda - 1 } } &= \frac { 1 } { { L - K -
\lambda } } \sum \nolimits_ { i = 1 } ^ { L - K - \lambda } \,
\,H_ { i , i + \lambda } ^ { \Lambda - 1 } \\ { \tau _ { \Lambda
\lambda } } &= \frac { 1 } { { L - K - \lambda } } \sum
\nolimits_ { i = 1 } ^ { L - K - \lambda } \, \,H_ { i , i +
\lambda } ^ \Lambda \\ \tag { 10 }
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Lambda$$
\end{document} represents the number of nucleotide physical properties and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${d_i}{ \rm{ \;}}$$
\end{document} represents the structural relationship of the \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\lambda-{ \rm{th}}$$
\end{document} layer of the nucleotide sequence:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\left\{ { \begin{matrix} {H_{i , i + j}^v = {P_v} \left( {{R_i}{R_{i + 1}}} \right) \cdot {P_v} \left( {{R_{i + j}}{R_{i + j + 1}}} \right) \; \; \; \; \; \; \; \; \; \; \; \; \; \; \; \; \; \; \; \; \; \;} \\ {v = 1 , 2 ,.. , \Lambda; j = 1 , 2 , \ldots , \lambda; i = 1 , 2 , \ldots , L - K - \lambda } \\ \end{matrix} } \right. , \tag{11}
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_v} \left( {{R_i}{R_{i + 1}}} \right)$$
\end{document} represents the value of the physical properties of the v nucleotides of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${R_i}{R_{i + 1}}$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${R_{i + j}}{R_{i + j + 1}}$$
\end{document}.
In general, the spatial position between the two base pairs can be described by six parameters, including three translation parameters and three rotation parameters. The three rotation parameters are, respectively, twist \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_1} \left( {{R_i}{R_{i + 1}}} \right)$$
\end{document}, tilt \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_2} \left( {{R_i}{R_{i + 1}}} \right)$$
\end{document}, and roll \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_3} \left( {{R_i}{R_{i + 1}}} \right)$$
\end{document}, and the three translation parameters are, respectively, shift \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_4} \left( {{R_i}{R_{i + 1}}} \right)$$
\end{document}, slide \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_5} \left( {{R_i}{R_{i + 1}}} \right)$$
\end{document}, and rise \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${P_6} \left( {{R_i}{R_{i + 1}}} \right)$$
\end{document}, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\left( {{R_i}{R_{i + 1}}} \right)$$
\end{document} represents the 16 possible dinucleotides AA, AC, AG, AU, …, UU (Chen et al., 2013). It should be noted that the data should be normalized:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{ P_v } \left( { { R_i } { R_ { i + 1 } } } \right) = { \frac { { P_v } \left( { { R_i } { R_ { i + 1 } } } \right) - \left\langle { { P_v } \left( { { R_i } { R_ { i + 1 } } } \right) } \right\rangle } { SD \left\langle { { P_v } \left( { { R_i } { R_ { i + 1 } } } \right) } \right\rangle } } , \tag { 12 }
\end{align*}
\end{document}
where < > represents the mean and SD represents the standard deviation. For the oligonucleotides, the values of original physicochemical property can be obtained from previous studies (Brukner et al., 1995; Chen et al., 2015). The six structural properties of RNA and their corresponding standard-converted values are given in Table 2.
The Six Structural Properties of RNA
Dinucleotide
Twist P1 (RiRi+1)
Tilt P2 (RiRi+1)
Roll P3 (RiRi+1)
Shift P4 (RiRi+1)
Slide P5 (RiRi+1)
RiseP6 (RiRi+1)
GG
0.347 (32)
−0.211 (0.3)
1.652 (12.1)
−0.551 (−0.01)
−1.407 (−1.78)
0.802 (3.32)
GA
0.347 (32)
1.321 (1.3)
0.413 (9.4)
0.147 (0.07)
−0.969 (−1.7)
1.515 (3.38)
GC
2.2 (35)
−0.67 (0)
−1.102 (6.1)
0.147 (0.07)
0.729 (−1.39)
0.386 (3.22)
GU
0.347 (32)
0.555 (0.8)
−1.698 (4.8)
1.545 (0.23)
0.51 (−1.43)
0.149 (3.24)
AG
0.888 (30)
0.096 (0.5)
0 (8.5)
−0.813 (−0.04)
0.127 (−1.5)
0.564 (3.3)
AA
−0.27 (31)
−1.896 (−0.8)
−0.689 (7)
−1.163 (−0.08)
1.386 (−1.27)
0.862 (3.18)
AC
0.347 (32)
0.555 (0.8)
−1.698 (4.8)
1.545 (0.23)
0.51 (−1.43)
0.149 (3.24)
AU
0.965 (33)
1.015 (1.1)
−0.643 (7.1)
−0.988 (−0.06)
0.893 (−1.36)
0.149 (3.24)
CG
2.741 (27)
−0.823 (−0.1)
1.652 (12.1)
2.156 (0.3)
−2.009 (−1.89)
0.564 (3.3)
CA
−0.27 (31)
0.862 (1)
0.643 (9.9)
0.497 (0.11)
0.346 (−1.46)
1.931 (3.09)
CC
0.347 (32)
−0.211 (0.3)
0.092 (8.7)
−0.551 (−0.01)
−1.407 (−1.78)
0.802 (3.32)
CU
0.888 (30)
0.096 (0.5)
0 (8.5)
−0.813 (−0.04)
0.127 (−1.5)
0.564 (3.3)
UG
−0.27 (31)
0.862 (1)
0.643 (9.9)
0.497 (0.11)
0.346 (−1.46)
1.931 (3.09)
UA
0.347 (32)
−0.977 (−0.2)
1.01 (10.7)
−0.639 (−0.02)
0.401 (−1.45)
0.089 (3.26)
UC
0.347 (32)
1.321 (1.3)
0.413 (9.4)
0.147 (0.07)
−0.969 (−1.7)
1.515 (3.38)
UU
−0.27 (31)
−1.896 (−0.8)
−0.689 (7)
−1.163 (−0.08)
1.386 (−1.27)
0.862 (3.18)
The values in parentheses are original data, and those outside are the standard converted value.
2.6. mRMR-based feature selection
Feature selection is a process to select the most effective features from a set of features to reduce the feature space dimension, and it is one of the key problems of pattern recognition (Zou et al., 2016b; Tang et al., 2017). Feature selection can save the computation time, reduce the requirement of measurement and storage, avoid over-fitting, and improve the prediction performance. In this study, mRMR method (Peng et al., 2005) was employed to pick out optimal features.
mRMR algorithm is a method to obtain the maximum correlation features and remove the redundant features. It takes into account not only the correlation between the feature and the label, but also the correlation between features. Metrics are mutual information and measure the dependency. mRMR can be considered as an approximation of the dependency relationship between the joint distribution of the maximized feature subset and the target variable:
Maximum relevance. The relevance of features and categories is the highest:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\max D \left( { S , c } \right) , { \rm { \; \; } } D = \frac { 1 } { { \left\vert s \right\vert } } \mathop \sum \limits_ { { x_i } \in S } I \left( { { x_i } ;c } \right). \tag { 13 }
\end{align*}
\end{document}
Minimum redundancy. The minimum redundancy between features
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\min R \left( { S , c } \right) , { \rm { \; \; } } R = \frac { 1 } { { { { \left\vert s \right\vert } ^2 } } } \mathop \sum \limits_ { { x_i } , { x_j } \in S } I \left( { { x_i } ; { x_j } } \right). \tag { 14 }
\end{align*}
\end{document}
In Equations (13) and (14), S represents the subset of features that have been chosen, c is class label, and x represents the feature. The final selection criteria are
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{ \rm{max}} \emptyset \left( {D , R} \right) , \emptyset = D - R. \tag{15}
\end{align*}
\end{document}
The resulting subset can ensure that the correlation between the feature and the category is maximum and that the redundancy of the feature is minimal.
2.7. Performance evaluation
To measure the prediction quality, the following four metrics: sensitivity (Sn), specificity (Sp), overall accuracy (Acc), and Matthews correlation coefficient (MCC) were used in this study (Feng et al., 2013a,b; Lin et al., 2014; Zhu et al., 2015; Jia et al., 2018). These measures are defined as follows:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\left\{ { \begin{matrix} {Sn = 1 - {{N_ - ^ + } \over {{N^ + }}}} & {0 \le Sn \le 1} \\ {Sp = 1 - {{N_ + ^ - } \over {{N^ - }}}} & {0 \le Sp \le 1} \\ {Acc = 1 - {{N_ - ^ + + N_ + ^ - } \over {{N^ + } + {N^ - }}}} & {0 \le Acc \le 1} \\ \displaystyle{MCC = {{1 - \Big( {{N_ - ^ + } \over {{N^ + }}} + {{N_ + ^ - } \over {{N^ - }}}\Big) } \over { \sqrt { \Big( 1 + {{N_ + ^ - - N_ - ^ + } \over {{N^ + }}} \Big) \Big(1 + {{N_ - ^ + - N_ + ^ - } \over {{N^ - }}} \Big) } }}} & {0 \le MCC \le 1} \\ \end{matrix} } \right. , \tag{16}
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${N^ + }$$
\end{document} is the total number of the 2′-O-methylation site sequences investigated, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N_ - ^ +$$
\end{document} is the number of 2′-O-methylation site sequences incorrectly predicted as the non-2′-O-methylation site sequence, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${N^ - }$$
\end{document} is the total number of the non-2′-O-methylation site sequences investigated, and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$N_ + ^ - { \rm{ \;}}$$
\end{document} is the number of the non-2′-O-methylation site sequences incorrectly predicted as the 2′-O-methylation site sequence.
Sn and Sp reflect the ability to correctly identify 2′-O-methylation sites and correctly recognize non-2′-O-methylation sites, respectively. Acc is the overall accuracy of the discrimination between 2′-O-methylation sites and non-2′-O-methylation sites. MCC values can intuitively measure a binary classification problem.
Meanwhile, the receiver operating characteristic (ROC) curve was also used to measure the prediction performance of the current method. Its vertical coordinate represents the true positive rate (sensitivity) and the horizontal coordinate represents the false positive rate. The area under the ROC curve, called AUC, is an objective index to evaluate a predictor. An AUC value of 0.5 is equivalent to random prediction and an AUC value of 1 represents a perfect one.
3. Results and Discussion
3.1. Cross-validation
Cross-validation is a commonly used statistical analysis method for objectively evaluating the performance of a classification model.
There are three commonly used cross-validation methods, independent data set test, n-fold cross-validation test, and jackknife cross-validation test (Chou, 2011). To save computational time, the fivefold cross-validation test was used to evaluate the performance of the proposed method in the study.
3.2. Parameter optimization
It can be seen from Equations (8) to (12) that there are three parameters to be optimized in our prediction model: \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\omega$$
\end{document}, k, and λ. Parameter ω represents the weight, and its value range is 0 to 1. Parameter k represents the short-range information of the RNA sequence and λ represents the long-range information of the sequence. It is obvious that the larger the values of k and λ are, the more the features were extracted. If the two values can be arbitrarily increased, it will usually cause excessive fitting and high-dimensional disaster and then decrease the accuracy of the prediction. Here, the optimal parameters are obtained through grid search as follows:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\left\{ { \begin{matrix} {2 \le k \le 6 , \; \; \hfill \Delta = 1\hfill} \\ {1 \le \lambda \le 5 , \; \; \hfill \Delta = 1\hfill} \\ {0 \le \omega \le 1 , \; \; \hfill \Delta = 0.1\hfill}\end{matrix} } \right.. \tag{17}
\end{align*}
\end{document}
Thus, we finally obtained 275 (5 × 5 × 11) results. We found that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\omega = 1$$
\end{document}, k = 3, and λ = 2 were the optimal values and produce the highest overall accuracy of 95.57%.
3.3. Feature selection results
We encoded RNA sequence by integrating the three nucleotide chemical properties, nucleotide composition, and type 2 PseKNC. Therefore, each sample in the benchmark data set was encoded by a 240-dimensional vector (3 × 41 + 41 + (43 + 6 × 2) = 240).
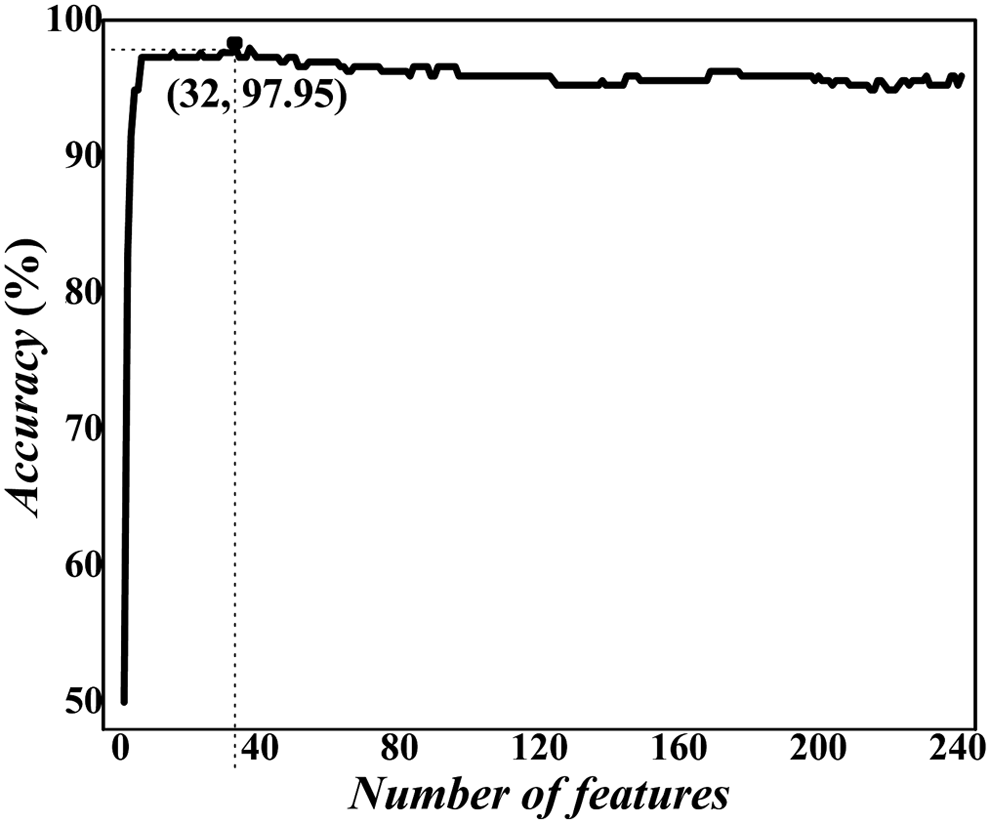
According to previous descriptions, to construct a robust and credible model, it is necessary to screen out key features from the 240 feature vectors. The mRMR method was used to rank the features. By adding the ranked features one by one according to the evaluations from mRMR, we established 240 SVM-based predictors. We then tested the prediction performance for the predictors and plotted the incremental feature selection curve as shown in Figure 2. When the top ranked 32 features were used, the maximum overall accuracy in the fivefold cross-validation test reached 97.95%. We also investigated the model's performance using jackknife cross-validation. Results showed that the overall accuracy of 97.62% was obtained, which was almost as high as that in fivefold cross-validated results. Interestingly, the 32 optimal features are all chemical properties, indicating that chemical properties play an important role in the prediction of 2′-O-methylation sites, whereas the nucleotide composition and type 2 PseKNC exist as noise. The principle of mRMR is to investigate whether there is correlation redundancy between features. Thus, we may conclude that sequence information and PseKNC cannot provide extra information to chemical properties for prediction. The chemical features are sufficient to describe the prediction problem of 2′-O-methylation sites.
A plot showing the incremental feature selection (IFS) procedure for identifying 2′-O-methylation site. When the top 32 features were used to perform prediction, the overall success rate reaches an IFS peak of 97.95% in fivefold cross-validation. IFS.
The detailed predictive performance of the predictors constructed from these 32 optimal features is expressed as follows:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
\left\{ { \begin{matrix} {Sn = 97.27 \% \; \;} \\ {Sp = 98.63 \% \; \;} \\ {Acc = 97.95 \% } \\ {MCC = 0.959 \; \;} \\ \end{matrix} } \right.. \tag{18}
\end{align*}
\end{document}
We also drew the ROC curve as shown in Figure 3. The AUC reached 0.9955, indicating that the proposed method was a promising high-throughput tool for predicting 2′-O-methylation sites. Then, we compared it with the previous method (Chen et al., 2016b) and found that the feature screening could dramatically improve the accuracy (Table 3).
The receiver operating characteristic curve for identifying 2′-O-methylation sites.
In the process of modeling, a suitable classification method is important for fast and reliable model construction. Neural network has been widely used in pattern recognition, especially for deep learning (Cao et al., 2016, 2017a,b). However, deep learning is more suitable for high-dimensional and large-sized samples. Moreover, it consumes more time and computational resources for optimizing model parameters. However, SVM is suitable for modeling small samples. Furthermore, the accuracy of SVM is high enough.
In SVM, four kernel functions (linear kernel, polynomial kernel, RBF kernel, and sigmoid kernel) have been used in classification. RBF kernel function has been widely used in biological data classification because it can map a sample into a higher dimensional space. However, it is necessary to investigate the performances of different kernel functions for comparison. Table 4 gives the prediction results of four kernel functions. Four kernel functions produced high prediction accuracies (>95%), demonstrating that the optimized features could reflect the intrinsic characteristics of 2′-O-methylation. Among the four kernel functions, RBF produced the maximum accuracy. Thus, the final model was established based on the RBF kernel function.
Comparison with Kernel Functions of Support Vector Machine
Kernel function
Feature selected technique
Acc (%)
Linear kernel
mRMR
97.27
Polynomial kernel
mRMR
96.59
RBF kernel
mRMR
97.95
Sigmoid kernel
mRMR
97.27
RBF, radial basis function.
3.4. Feature analysis
The mentioned results showed that our model could produce high accuracy. However, we also noticed that eight samples were not correctly predicted. Thus, we should further investigate why these samples were not correctly identified. At first, we statistically analyzed the distribution difference of the 32 optimal features between positive and negative samples with Equation (19).
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{ u_i } = \frac { { f_i^P - f_i^N } } { S } , \tag { 19 }
\end{align*}
\end{document}
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$f_i^P$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\usepackage{upgreek}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$f_i^N$$
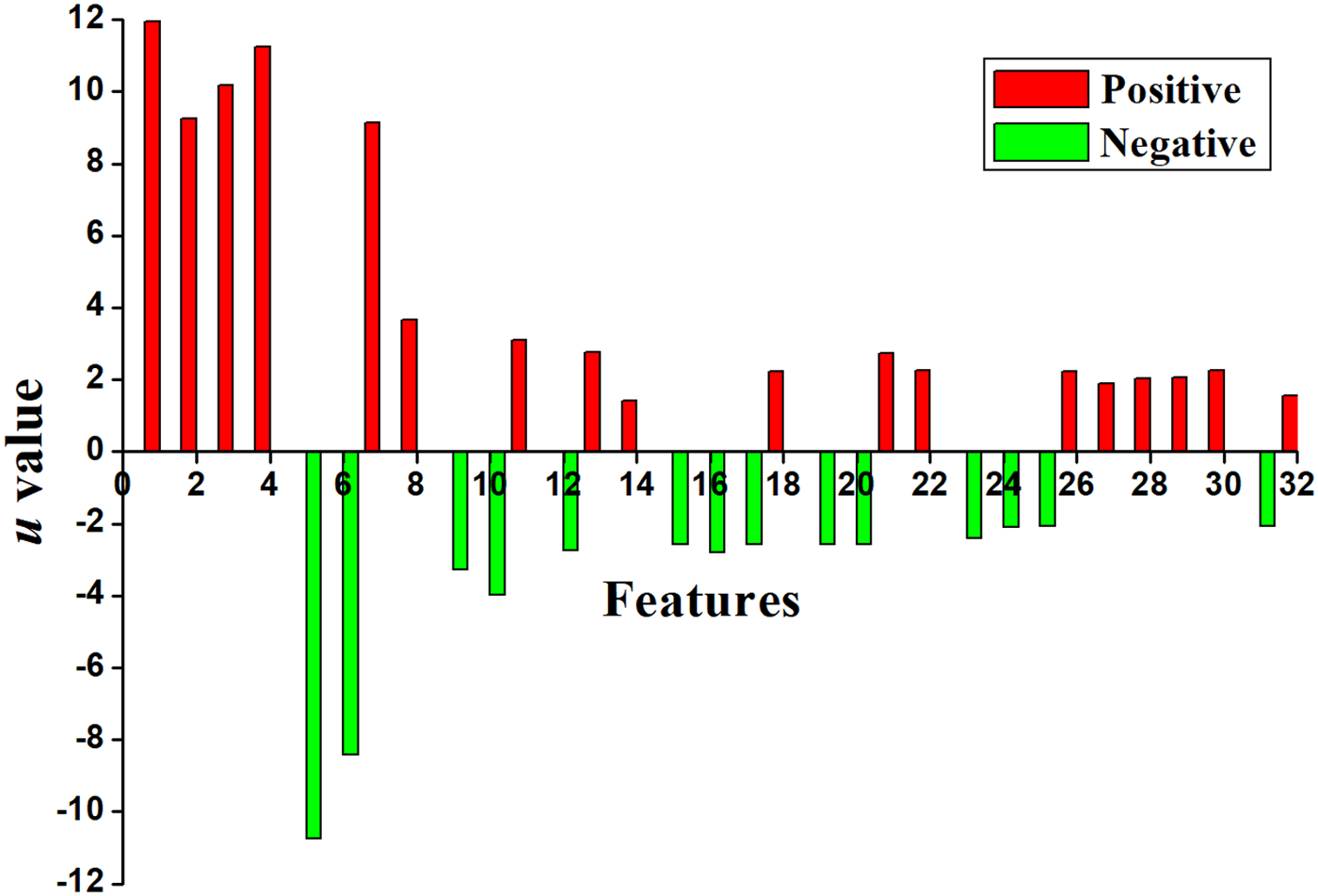
\end{document}, respectively, denote the occurrence frequencies of the i-th feature (i = 1, 2, …, 32) in positive and negative samples. The denominator S is a standard error. The 32 u values from 32 optimal features are plotted in Figure 4. Red (green) bars indicate that the features prefer to occur in positive (negative) samples.
A chromaticity diagram for the 32 optimal features. The red bars indicate that the features prefer positive samples, whereas green bars indicate the features prefer negative samples.
The eight incorrectly predicted samples including six incorrectly positive samples and two incorrectly negative samples. Further statistical results showed that the features of the six negative samples always occurred in positive samples. For example, all of them contain first four features (red) and excluded the fifth and sixth features. The two positive samples possessed the features that usually occurred in negative samples. Therefore, the eight samples were incorrectly predicted.
3.5. Web server guide
A web server can provide convenience for experimental scholars (Liang et al., 2017; Zhang et al., 2017). Thus, a novel platform obtained through the mentioned procedures has been developed and named “iRNA-2OM,” in which “i” means identify, “RNA” means the samples of RNA sequences, and “2OM” means 2′-O-methylation. A step-by-step guide of the web server is given as follows:
Step 2. Click on the WEBSERVER button in banner and type or copy/paste the query RNA sequences into the input box at the center. The input sequence should be in the FASTA format. Example sequences in FASTA format can be obtained by clicking the example button (Fig. 5).
Step 3. Click on the SUBMIT button to see the predicted result. You will see the outcomes shown on the screen of your computer.
Step 4. Click on the CITATION button to find the relevant articles that document the detailed development and algorithm. The DOWNLOAD button provides the benchmark data sets used in our model. By clicking on the HELP button, readers can read the relevant instructions and the caveat for use. The CONTACT button provides relevant information about the developer.
2′-O-methylation plays critical roles in regulating gene expressions at the post-transcriptional levels. Thus, proper identification of the 2′-O-methylation site is crucial to the understanding of the mechanism of RNA. In this study, we proposed a SVM-based model to predict 2′-O-methylation sites in H. sapiens. The RNA sequence samples were encoded by nucleotide chemical properties, nucleotide composition, and type 2 PseKNC. The mRMR was used to pick out the optimal features. In the fivefold cross-validation test, an accuracy of 97.95% was obtained. Comparison with other published methods showed that the proposed method is superior to other methods. Based on the method, we established a free predictor called iRNA-2OM that can be freely accessible at (http://lin-group.cn/server/iRNA-2OM/). We hope that our method will become a useful tool for identifying 2′-O-methylation sites.
Footnotes
Acknowledgments
This work was supported by the National Nature Scientific Foundation of China (Grant Nos. 61772119 and 31771471), the Natural Science Foundation for Distinguished Young Scholar of Hebei Province (Grant No. C2017209244), the Fundamental Research Funds for the Central Universities of China (Grant Nos. ZYGX2015Z006, ZYGX2016J118, ZYGX2016J125, and ZYGX2016J223), program for the Top Young Innovative Talents of Higher Learning Institutions of Hebei Province (Grant No. BJ2014028), the Outstanding Youth Foundation of North China University of Science and Technology (Grant No. JP201502), and China Postdoctoral Science Foundation (Grant No. 2015M582533).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
1.
BachellerieJ.P., CavailleJ., and HuttenhoferA.2002. The expanding snoRNA world. Biochimie, 84, 775–790.
2.
BruknerI., SanchezR., SuckD., et al.1995. Sequence-dependent bending propensity of DNA as revealed by DNase I: Parameters for trinucleotides. EMBO J, 14, 1812–1818.
3.
CaoR., FreitasC., ChanL., et al.2017a. ProLanGO: Protein function prediction using neural machine translation based on a recurrent neural network. Molecules, 22, 1732.
4.
CaoR., WangZ., WangY., et al.2014. SMOQ: A tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC Bioinformatics, 15, 120.
5.
CaoR.Z., AdhikariB., BhattacharyaD., et al.2017b. QAcon: Single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics, 33, 586–588.
6.
CaoR.Z., BhattacharyaD., HouJ., et al.2016. DeepQA: Improving the estimation of single protein model quality with deep belief networks. BMC Bioinformatics, 17, 495.
7.
ChangC.C., HsuC.W., and LinC.J.2000. The analysis of decomposition methods for support vector machines. IEEE Trans Neural Netw, 11, 1003–1008.
8.
ChenW., FengP., DingH., et al.2016a. PAI: Predicting adenosine to inosine editing sites by using pseudo nucleotide compositions. Sci. Rep., 6, 35123.
9.
ChenW., FengP., TangH., et al.2016b. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics, 107, 255–258.
10.
ChenW., FengP., TangH., et al.2016c. RAMPred: Identifying the N(1)-methyladenosine sites in eukaryotic transcriptomes. Sci. Rep., 6, 31080.
11.
ChenW., FengP.M., LinH., et al.2013. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 41, e68.
12.
ChenW., LeiT.Y., JinD.C., et al.2014. PseKNC: A flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem., 456, 53–60.
13.
ChenW., YangH., FengP., et al.2017. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics, 33, 3518–3523.
14.
ChenW., ZhangX., BrookerJ., et al.2015. PseKNC-general: A cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics, 31, 119–120.
15.
ChenX.X., TangH., LiW.C., et al.2016d. Identification of bacterial cell wall lyases via pseudo amino acid composition. Biomed. Res. Int., 2016, 1654623.
16.
ChouK.C.2011. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol., 273, 236–247.
17.
DaoF.Y., YangH., SuZ.D., et al.2017. Recent advances in conotoxin classification by using machine learning methods. Molecules, 22, 1057.
18.
DecaturW.A., and FournierM.J.2002. rRNA modifications and ribosome function. Trends Biochem. Sci., 27, 344–351.
19.
DongZ.W., ShaoP., DiaoL.T., et al.2012. RTL-P: A sensitive approach for detecting sites of 2′-O-methylation in RNA molecules. Nucleic Acids Res. 40, e157.
20.
FengP., DingH., YangH., et al.2017. iRNA-PseColl: Identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Mol. Ther. Nucleic Acids, 7, 155–163.
21.
FengP., YangH., DingH., et al.2018. iDNA6mA-PseKNC: Identifying DNA N(6)-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics. [Epub ahead of print]; DOI:10.1016/j.ygeno.2018.01.005.
22.
FengP.M., DingH., ChenW., et al.2013a. Naive Bayes classifier with feature selection to identify phage virion proteins. Comput. Math. Methods Med., 2013, 530696.
23.
FengP.M., LinH., and ChenW.2013b. Identification of antioxidants from sequence information using naive Bayes. Comput. Math. Methods Med., 2013, 567529.
24.
FuL., NiuB., ZhuZ., et al.2012. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–3152.
25.
JiaC., ZuoY., ZouQ., et al.2018. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics. [Epub ahead of print]; DOI:10.1093/bioinformatics/bty039.
26.
KissT.2002. Small nucleolar RNAs: An abundant group of noncoding RNAs with diverse cellular functions. Cell, 109, 145–148.
27.
LaiH.Y., ChenX.X., ChenW., et al.2017. Sequence-based predictive modeling to identify cancerlectins. Oncotarget, 8, 28169–28175.
28.
LiJ., YangZ., YuB., et al.2005. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol., 15, 1501–1507.
29.
LiangZ.Y., LaiH.Y., YangH., et al.2017. Pro54DB: A database for experimentally verified sigma-54 promoters. Bioinformatics, 33, 467–469.
30.
LinH., DengE.Z., DingH., et al.2014. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 42, 12961–12972.
31.
LinH., LiangZ.Y., TangH., et al.2017. Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. [Epub ahead of print]; DOI:10.1109/TCBB.2017.2666141.
32.
MadenB.E.2001. Mapping 2′-O-methyl groups in ribosomal RNA. Methods, 25, 374–382.
33.
ManavalanB., BasithS., ShinT.H., et al.2017. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget, 8, 77121–77136.
34.
ManavalanB., and LeeJ.2017. SVMQA: Support-vector-machine-based protein single-model quality assessment. Bioinformatics, 33, 2496–2503.
35.
ManavalanB., ShinT.H., and LeeG.2018a. DHSpred: Support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget, 9, 1944–1956.
36.
ManavalanB., ShinT.H., and LeeG.2018b. PVP-SVM: Sequence-based prediction of phage virion proteins using a support vector machine. Front. Microbiol., 9, 476.
37.
PengH., LongF., and DingC.2005. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern. Anal. Mach. Intell., 27, 1226–1238.
38.
RamachandranV., and ChenX.2008. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science, 321, 1490–1492.
39.
SchölkopfB., Burges, ChristopherJ.C., and SmolaA.J. (eds). 1999. Advances in Kernel Methods: Support Vectror Learning. MIT Press, Cambridge, MA.
40.
SunW.J., LiJ.H., LiuS., et al.2016. RMBase: A resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 44, D259–D265.
41.
TangH., CaoR.Z., WangW., et al.2017. A two-step discriminated method to identify thermophilic proteins. Int. J. Biomath. 10, 1750050.
42.
TangH., ChenW., and LinH.2016. Identification of immunoglobulins using Chou's pseudo amino acid composition with feature selection technique. Mol. Biosyst., 12, 1269–1275.
43.
YangH., TangH., ChenX.X., et al.2016. Identification of secretory proteins in Mycobacterium tuberculosis using pseudo amino acid composition. Biomed. Res. Int. 2016, 5413903.
44.
ZhangT., TanP., WangL., et al.2017. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 45, D135–D138.
45.
ZhaoY.W., LaiH.Y., TangH., et al.2016. Prediction of phosphothreonine sites in human proteins by fusing different features. Sci. Rep. 6, 34817.
46.
ZhaoY.W., SuZ.D., YangW., et al.2017. IonchanPred 2.0: A tool to predict ion channels and their types. Int J Mol Sci, 18, E1838.
47.
ZhuP.P., LiW.C., ZhongZ.J., et al.2015. Predicting the subcellular localization of mycobacterial proteins by incorporating the optimal tripeptides into the general form of pseudo amino acid composition. Mol. Biosyst., 11, 558–563.
48.
ZouQ., JuY., and LiD.2016a. Protein folds prediction with hierarchical structured SVM. Curr. Proteomics, 13, 79–85.
49.
ZouQ., ZengJ., CaoL., et al.2016b. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing, 173, 346–354.
50.
ZustR., Cervantes-BarraganL., HabjanM., et al.2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol., 12, 137–143.