
Abstract
Abstract
Typhoid fever is an acute illness in humans, caused by Salmonella typhi, a gram-negative bacterium. Outer membrane proteins of S. typhi have strong potential for its use in the development of subunit vaccine against typhoid. In the current study, peptide-based subunit vaccine was constructed from outer membrane protease E (PgtE) against S. typhi. B cell and T cell epitopes were identified at fold level with a validated three-dimensional modeled structure. T cell epitopes from PgtE (IHPDTSANY) have 99.5% binding to a maximum number of major histocompatibility complex class I and class II alleles. They also bind to the typhoid-resistant human leukocyte antigen (HLA) alleles DRB1*0401. PgtE epitopes were docked with HLA-DR4 (PDB ID: 1D5M) and a contact map was constructed. A simulation search for the binding site for full flexibility of the peptide from CABS- (Cα, Cβ, side-chain)-dock shows stable interactions. Molecular dynamics simulation studies revealed that the PgtE-epitope complex structure was more stable throughout the simulation (20 ns) and interaction did not change the radius of gyration. In conclusion, computational analysis, molecular docking, and molecular dynamics (MD) simulation of PgtE-epitope complex were used to elucidate the binding mode, and the dynamical changes of epitopes were more suitable for vaccine development against typhoid.
1. Introduction
Salmonella typhi pathogen causes diseases in humans with severity varying from mild diarrhea to severe systemic infections such as typhoid fever. Typhoid fever remains a serious public health concern, with an estimated 22 million cases and 200,000 related deaths occurring worldwide each year (Crump et al., 2004; Hamid and Jain, 2008; Crump and Mintz, 2010; Mathur et al., 2012). In addition, it was estimated that up to 1.3 billion cases of acute gastroenteritis are caused, leading to 3 million deaths annually (Pang et al., 1995; Samykannu et al., 2017; Samykannu et al., 2018). Effective vaccination strategies are highly required against typhoid fever. Unfortunately, two currently available (Ty21a and parenteral Vi vaccine) approved vaccines have only modest efficacy (50%–80% protection) (Guzman et al., 2006). The outer membrane proteins (OMPs) of Salmonella have been considered potential candidates for conferring protection against typhoid (Luis Puente et al., 1987; Isibasi et al., 1988; Puente et al., 1989, 1991; Hamid and Jain, 2008; Samykannu et al., 2014). Thus, OMPs have a strong prospective for the development of a subunit vaccine against typhoid.
Peptide-based vaccine has the advantage of delivering high doses of potential immunogen at low-cost affordability to resource-poor countries (Nascimento and Leite, 2012; Rangasamy et al., 2018; Vijayababu et al., 2018). A whole protein-based antigen is not essentially required for eliciting protective response, while an immunodominant or epitope-rich segment of protein is sufficient to mount a protective immune response (Disis et al., 1996). Peptide-based subunit vaccine development has gained momentum recently due to its success in treating infectious diseases and promoting destruction of cancerous cell (Florea et al., 2003). Effective immune response-based clearance of pathogen in system depends on binding of epitope to human leukocyte antigen (HLA) alleles and major histocompatibility complex (MHC) class I (recognize CD8+ T cells) and MHC class II (recognize CD4+ T cells) molecules (Germain, 1994; Watts, 1997).
Activation of both helper T lymphocytes and cytotoxic T lymphocytes (CTL) requires recognition of specific peptides bound to MHC molecules on the antigen-presenting cells (APCs)/target cells. Protein antigens inside the APCs/target cells are degraded into small peptide fragments by the intracellular proteases. After partial proteolysis, some of the peptides, called T cell epitope, bind to MHC molecules and are transported to the cell surface for recognition by the antigen-specific T cell receptors. Thus, MHC binding is a prerequisite for a peptide to be a T cell epitope (Singh and Raghava, 2001). A peptide should lack a proteosomal recognition site if used as a vaccine candidate, otherwise, it degrades during antigen processing (Toes et al., 2001).
In this study, we have focused on designing a peptide-based subunit vaccine based on the OMP (PgtE) of S. typhi, which can elicit both B cell- and T cell-mediated immunity. Binding stability of these epitopes with typhoid fever resistance HLA-DR4 was analyzed.
2. Methods
2.1. Target protein retrieval and domain architecture
PgtE (Q8Z4Y4) amino acid sequence was retrieved from UniProtKB (Apweiler et al., 2004). The achievable domain characteristic included in PgtE was investigated by InterPro (Hunter et al., 2009).
2.2. Antigenicity prediction
PgtE amino acid sequence was subjected to VaxiJen v2.0 (Doytchinova and Flower, 2007) to predict antigenic property. This bioinformatic tool analyzed antigenic property based on transformation of protein sequences into uniform vectors of principal amino acid properties. The default parameter of threshold is 0.4 and anti-autocross-covariance output was used against full-length protein.
2.3. Prediction of B cell and T cell epitopes
PgtE amino acid sequence was subjected to BCPred. Antigenic linear nonoverlapping 20-mer B cell epitopes were predicted from whole antigen using BCPreds software with two methods. The amino acids pair scaling (AAP) method, based on amino acid pair antigenicity (Chen et al., 2007) and BCPreds, using the subsequence kernel provided by BCPreds, were followed to predict more reliable B cell epitope. Only B cell epitopes having a score >0.8 were accepted. Selected B cell epitopes were subjected to VaxiJen for antigenicity check. B cell epitopes having antigenic property, BCPreds score >0.8, and VaxiJen score >0.4 were selected for T cell epitope prediction.
Selected B cell epitopes were subjected to ProPred I (Singh and Raghava, 2003) and ProPred (Guan, 2003) analysis. ProPred predicted MHC class II binding peptides for 51 alleles and ProPred1 to predict MHC class I binding peptides (CTL epitopes) for 47 alleles. Common epitopes that bind to both the MHC classes were selected. The selected epitope was then analyzed with VaxiJen. IC50 values of corresponding epitopes were deduced from MHCPred server (Oprea and Antohe, 2013). IC50 < 50 nm a good binder, IC50 = 50–500 nm an intermediary binder, IC50 = 500–5000 nm a weak binder, and IC50 > 5000 a nonbinder. As the DRB1*0101 is the commonest bound allele, epitopes with IC50 value less than 100 nm were selected.
T-Epitope Designer (Kangueane and Sakharkar, 2005) was used for screening the HLA-peptide binding epitope based on virtual binding pockets using crystal structures of HLA-peptide complexes followed by calculation of peptide binding to binding pockets. The next screening criteria include the following: (1) capability of binding with a large number of HLA alleles (>1000), (2) must bind to DRB1*0101 and DRB1*0401, and (3) must bind to A*0201, A*0204, and A*2705 (Prabhavathy et al., 2011). The final list of epitopes was selected based on the abovementioned criteria and VaxiJen score. Selected epitopes were further analyzed for fold-level topology.
2.4. Three-dimensional modeling, refinement, and validation
I-TASSER, a hierarchical protein structure modeling approach based on the secondary structure enhanced profile/profile threading alignment (Roy et al., 2010), was used to model PgtE. The best model was selected based on the computed C-score. Modeled three-dimensional (3D) structures were subjected to ModRefiner (Xu and Zhang, 2011) for refinement of energy minimization. PROCHECK (Laskowski et al., 1993) was used to validate the refined 3D structure.
2.5. Fold-level topology analysis
The folding and clusters of selected epitopes in folded PgtE were analyzed to confirm the topology of epitope using Pepitope server (Mayrose et al., 2007), which uses PepSurf and Mapitope algorithms (Mayrose et al., 2006) to analyze the fold-level topology in PgtE.
2.6. Characterization of epitopes
Short sequence of predicted epitopes of PgtE was modeled using CABS- (Cα, Cβ, side-chain)-fold server (Blaszczyk et al., 2013). Secondary structures were analyzed, and resultant epitopes were subjected to ProGene 1.0 (Vijayababu et al., 2017a) to determine molecular weight and isoelectric point (pI).
2.7. Peptide docking
Identified epitopes from PgtE were docked with HLA-DR4 (PDB ID: 1D5M) using CABS-dock, which performs simulation search for the binding site permitting full flexibility of the peptide and small fluctuations of the receptor backbone (Kurcinski et al., 2015). These searches were used for favorable interactions between epitope and MHC molecule. Protein/peptide docking in CABS-dock is divided into three repeated steps realized by separate protocols: (1) prediction of the binding site(s) on the receptor structure, (2) initial modeling of the peptide backbone in the binding site(s), and finally, (3) refinement of the initial protein/peptide complexes to high resolution. Accuracy of the best obtained models was calculated based on the coordinate of root-mean-square deviation (Blaszczyk et al., 2016) between the epitope and HLA molecule.
2.8. Molecular dynamics simulation of receptor/ligand complex
To understand the dynamic behavior of HLA-DR4, PgtE epitope was subjected to molecular dynamics (MD) simulation for a time period of 20 ns using GROMACS 5.0.4 package (Van Der Spoel et al., 2005). Protein topology was obtained by GROMOS96 54a7 force field for apo and complexes of HLA-DR4. The docked complex was centered in a cubic box while the periodic boundary condition (PBC) condition was kept as 1 Å from each edge of the box. We used the simple point charge (SPC) water model to fill the surrounding of the protein or protein complexes within the box, and the system was neutralized by the addition of exact counter ions by replacing equal numbers of water molecules. A steepest descent method was applied for 50,000 steps with a maximum of 1000 kJ/mol/nm force to minimize the energy of the system, and the conjugate gradient was continued with the same parameter setup for the system. Van der Waals and long-range electrostatic interactions within the system were treated with particle mesh Ewald (Kawata and Mikami, 2001), while all covalent bonds with LINCS algorithm (Hess et al., 1997) and geometry of water with SETTLE algorithm (Miyamoto and Kollman, 1992). Moreover, the system was equilibrated by the isothermal/isochoric [or number of volume and temperature (NVT)] ensemble method with a temperature of 300 K using V-rescale method. Berendsen et al. (1984) further proceeded with [number of particles and temperature (NPT)] at a constant pressure of 1 atm using the Parrinello and Rahman method (Parrinello and Rahman, 1981) for a time period of 500 ps. With this, the whole system was subjected to MD simulation production for 20 ns using leapfrog integrator. The energy and coordinates of protein and protein complexes were stored in their corresponding trajectories at each 2 ps interval and analyzed with the Xmgrace tool. Overall workflow is shown in Figure 1.

Overall, workflow of PgtE epitope identification.
3. Results and Discussion
3.1. Domain prediction and antigenicity scoring
PgtE belongs to the outer membrane adhesion omptin domain and amino acid sequence ranging from 12 to 312 (InterPro number IPR020080), and antigenicity was analyzed using VaxiJen v2.0, assigning a threshold value of 0.4 for antigenic proteins and PgtE scored 0.6.
3.2. Identification of B cell epitope
Amino acid sequence of PgtE (312 aa) was subjected to BCPred for B cell epitope prediction. Both BCPred and AAP fixed length epitope prediction methods were used, and predicted B cell epitopes are listed in Tables 1 and 2. Epitope length was set as 20 and specificity 75%. Epitopes having BCPreds and VaxiJen scores >0.8 and >0.4, respectively, were selected. Finally, four of five epitopes from PgtE were selected for further analysis.
Predicted B Cell Epitopes from PgtE Using BCPred (BCPred Method)
Antigenicity of B cell epitopes of PgtE was calculated using VaxiJen. Selected B cell epitopes are in bold.
A, antigen; NA, nonantigen.
Predicted B Cell Epitopes from PgtE Using BCPred [Amino Acids Pair Scaling (AAP) Method]
Antigenicity of B cell epitopes of PgtE was calculated using VaxiJen. Selected B cell epitopes are in bold.
3.3. Identification of T cell epitope
Selected B cell epitopes from PgtE were subjected to ProPred1 and ProPred with default parameters for identification of T cell epitopes. PgtE epitopes were selected based on VaxiJen score, MHCPred score, and on surface localization of epitope in theoretical model. Common epitopes bind to a maximum number of both the MHC class I and II alleles and specifically interact with DRB1*0101 and DRB1*0401. IC50 values for both are listed in Table 3.
Epitopes That Can Induce Both the B Cell- and T Cell-Mediated Immunity Are Represented Along with Their Various Parameters
Identified epitopes with their binding abilities to >1000 MHC alleles using T-Epitope Designer. The epitopes that bind to >80% alleles were selected (Table 4). These epitopes also bind to DRB1*0401 alleles belonging to MHC class-II. The DRB1*0401 allele gives resistance against typhoid fever (Dunstan et al., 2000). A complex between this allele and selected epitope was subjected to interaction study.
T Cell Epitope Prediction Using T-Epitope Designer
IHPDTSANY has a bind to 99.5% of alleles—the lowest score with (A*3102) and highest score with (B*4104).
3.4. Modeling and structure validation
The modeling was done through I-TASSER server, where the modeled PgtE possessed a C-score of −0.1 (Vijayababu et al., 2017b). C-score is a confidence score for estimating the quality of predicted models by I-TASSER. It was calculated based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations. Structure refinement in ModRefiner shows PgtE (Fig. 2) with root-mean-square deviation (RMSD) of 0.650 and template modeling (TM) score is −0.9914 from an initial model. Ramachandran plot calculations were estimated through PROCHECK program (Supplementary Table S1 and Supplementary Fig. S1).
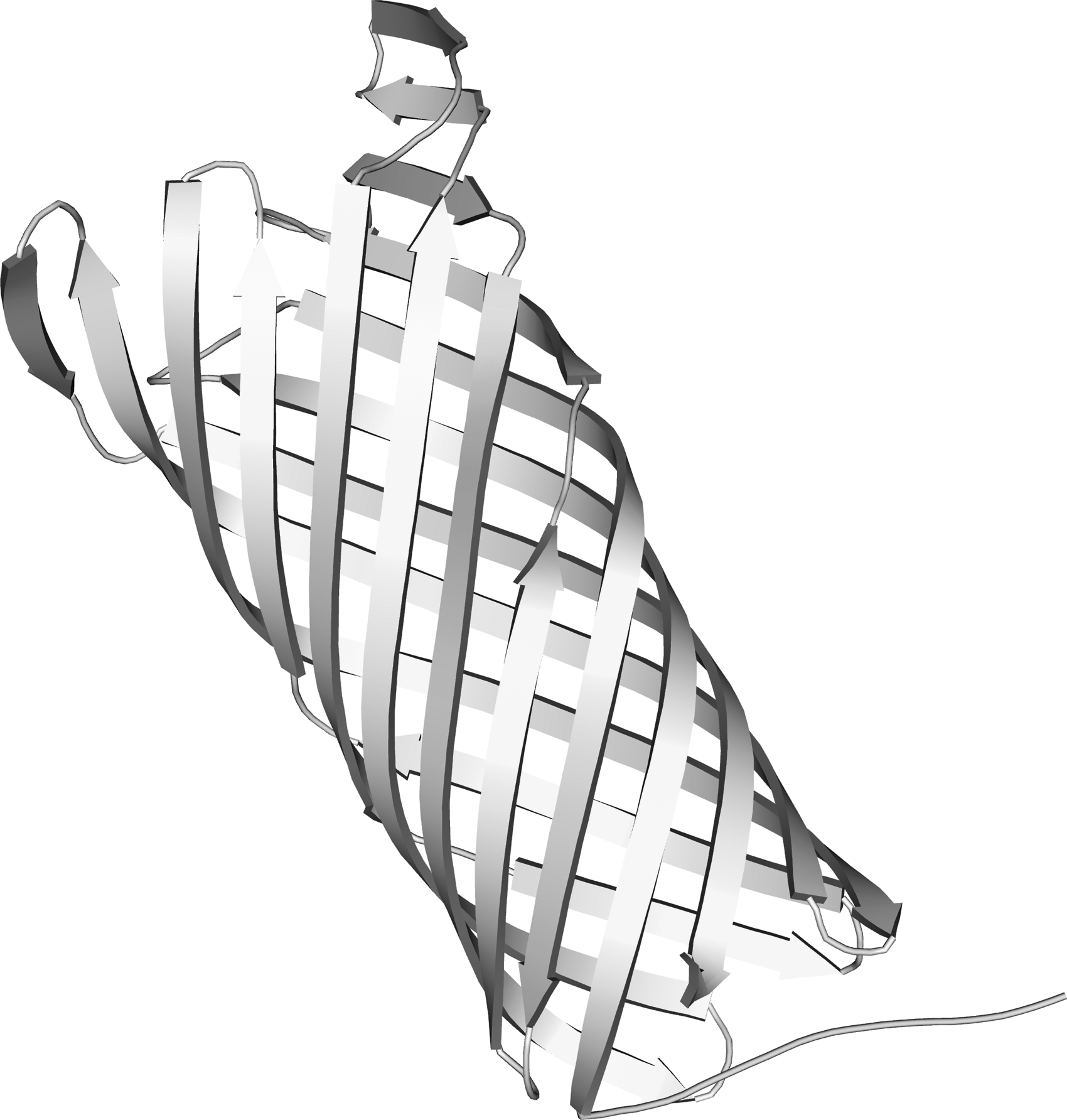
3D theoretical structures of PgtE. 3D, three dimensional.
3.5. Topology and characterization of T cell epitopes
The position of predicted epitopes on theoretical models of PgtE was identified using Pepitope server. Input requirement was epitope sequences and modeled structure of the antigen. The epitopes have also been found to bind selected MHC molecules (A*0201, B*2705, and DRB1*0401) and also bind to HLA molecules with 99.50% for PgtE in T-Epitope Designer.
The PgtE epitope “

Position of predicted epitopes shown on theoretical models of PgtE, differentiated by color.
3.6. Theoretical model of PgtE epitopes
The CABS-fold web server was used to generate three-dimensional structure of predicted epitopes by de novo modeling, shown in Figure 4. Molecular weight for PgtE epitope was calculated as 1017.06 Da. Theoretical isoelectric point (pI) calculated as 5.08.
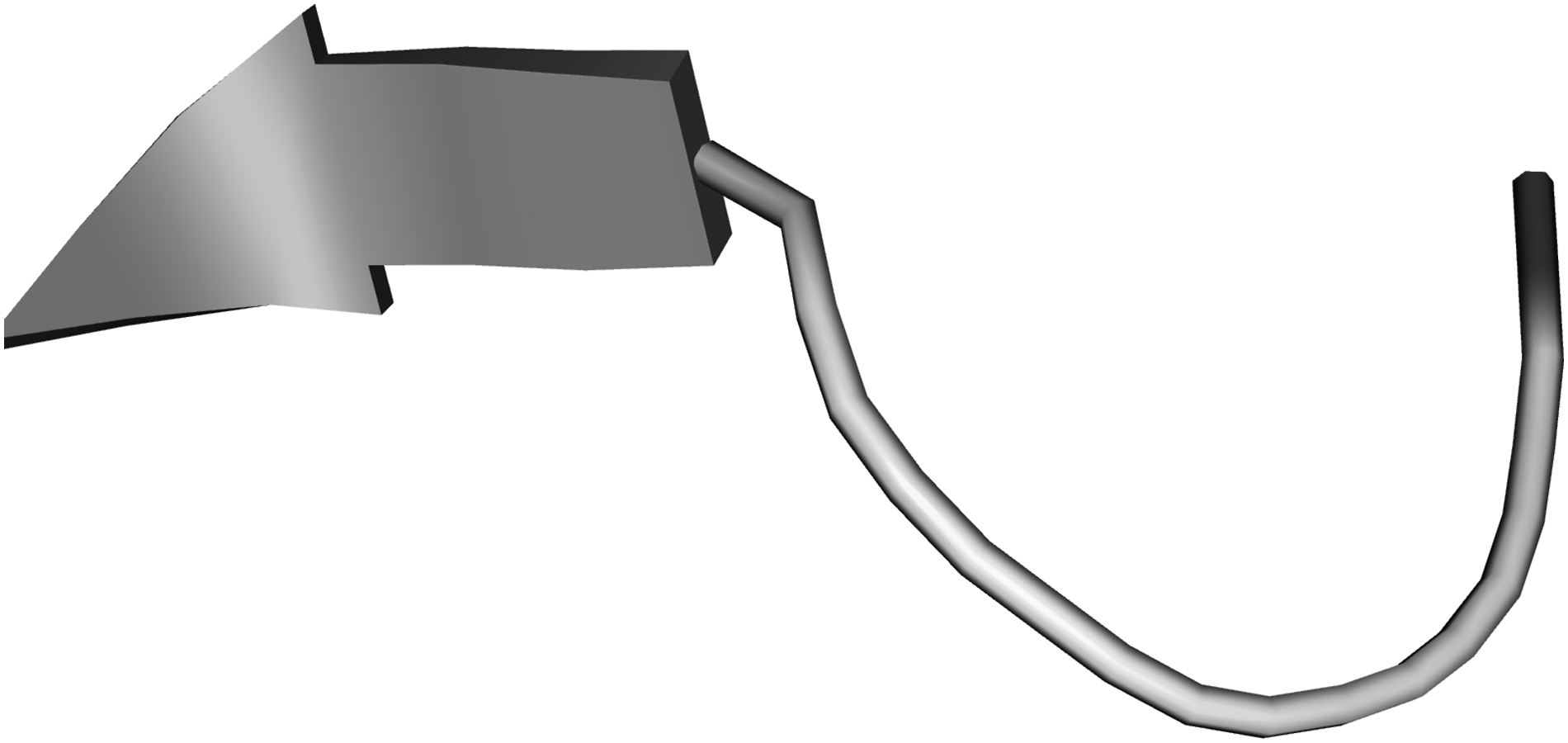
3D theoretical structure of PgtE (IHPDTSANY) epitope modeled using CABS-fold server. CABS, Cα, Cβ, side-chain.
3.7. Epitope docking with HLA-DR4 antigen
PgtE epitope was docked with CABS-dock web server and generates 10,000 model structures of the protein/peptide complex. Clustering based on the RMSD of the entire protein/epitope complex and resulting structures are grouped in clusters of similar complexes and ranked according to cluster size from the largest to the 10th largest. Ten top-ranked CABS-dock models, representatives of the 10 most numerous clusters, are shown in Supplementary Table S2, and the top-ranked docked model is shown in Figure 5.
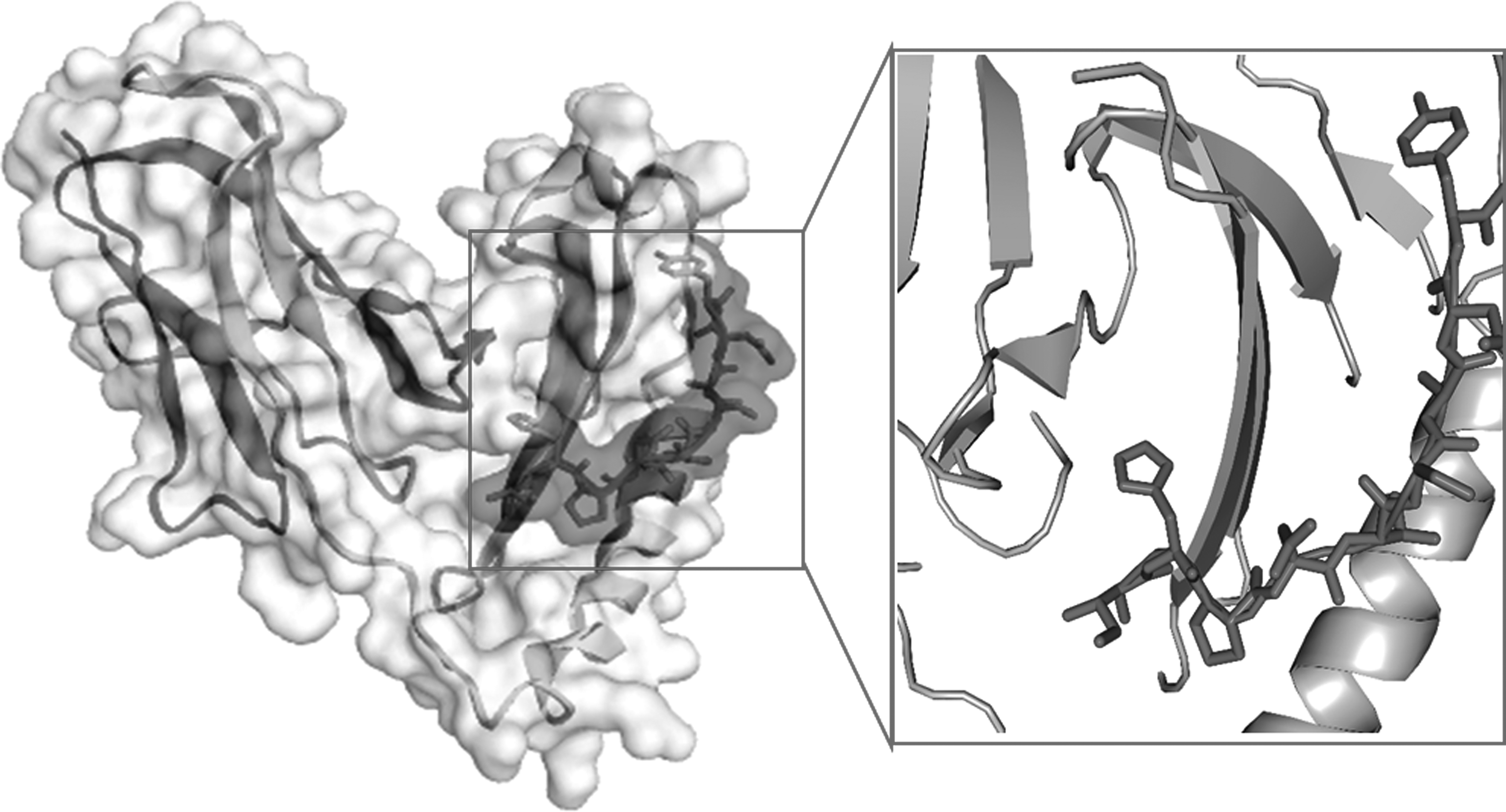
3D crystal structure of HLA class II histocompatibility antigen (PDB ID: 1D5M) docked with PgtE epitope (IHPDTSANY) represented as light grey stick. HLA, human leukocyte antigen.
3.8. Contact map analysis
The interactions of highly dynamic complexes from PgtE epitopes with HLA-DR4 can be analyzed using contact maps derived from the CABS-dock simulations. Intermolecular protein/peptide interactions were analyzed and the contact map is shown in Figure 6A, and contact residues are shown in Supplementary Table S3. We examined the contact-forming residues of the peptide to find out which parts of the PgtE modeling peptide are most important for its interaction with HLA-DR4. According to the interaction study, the most important complex-stabilizing contacts are formed by three PgtE (HIS 180, TYR 187, and ILE179), with the binding threshold as 4. In our docking results, we observed the highest contact frequencies for their neighbors as shown in histogram Figure 6B.

3.9. Molecular dynamic simulation
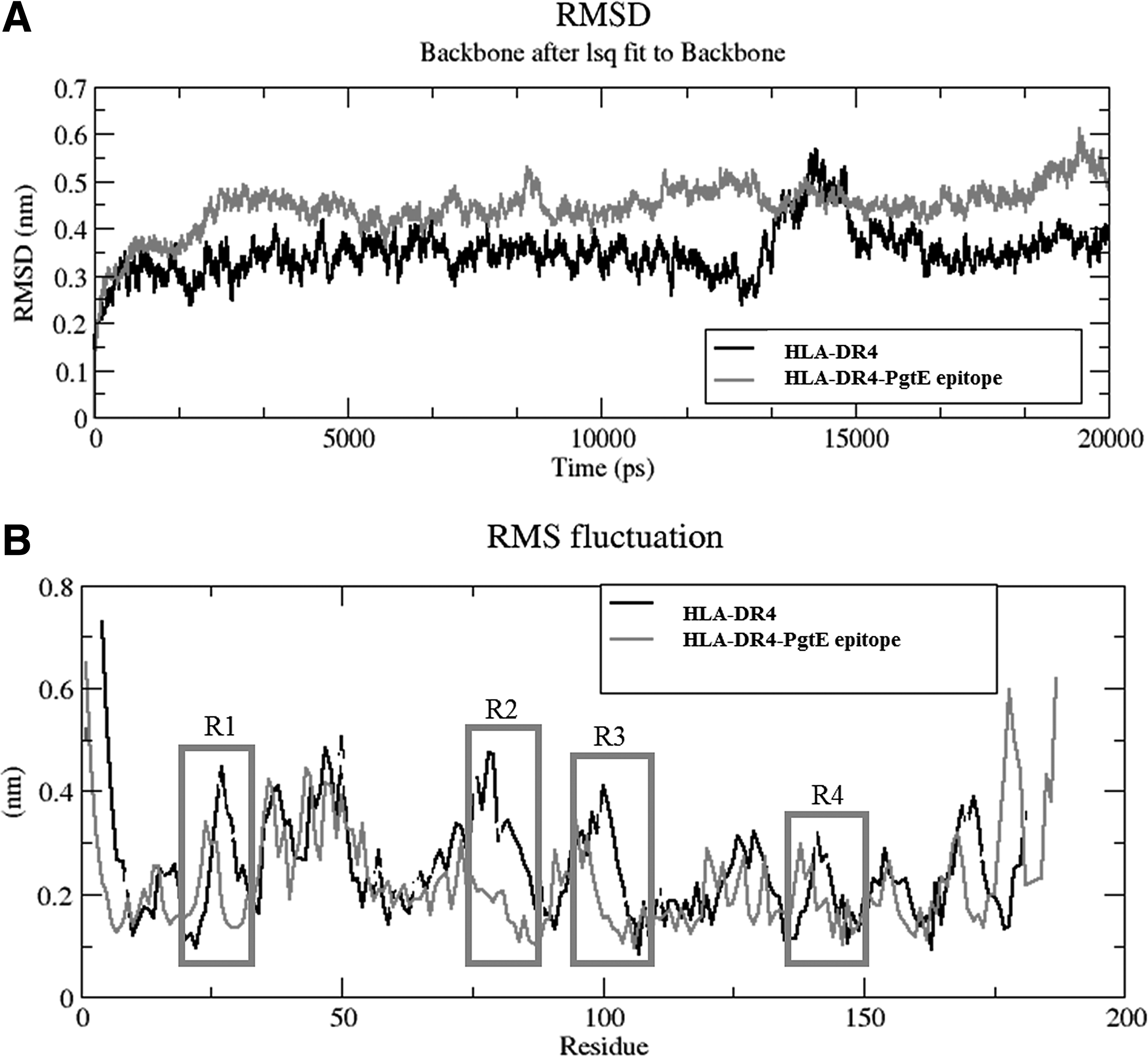
From MD simulation analysis, it was shown that apo from HLA-DR4 showed more RMSD fluctuations compared with PgtE epitope, except at 12.5 to 15 ns time period (Fig. 7A). The difference in RMSD was observed by large conformational changes in 14–19, 34–40, 58–77, 93–102, and 153–161 aa regions of HLA-DR4 (Fig. 7B). The epitope complex structure was more stable throughout the simulation, and PgtE interaction did not change the radius of gyration of apo and PgtE epitope bound form of HLA, where the Rg shuttles between 1.8 and 1.95 nm (Fig. 8A).
Molecular dynamics (MD) simulation analysis.
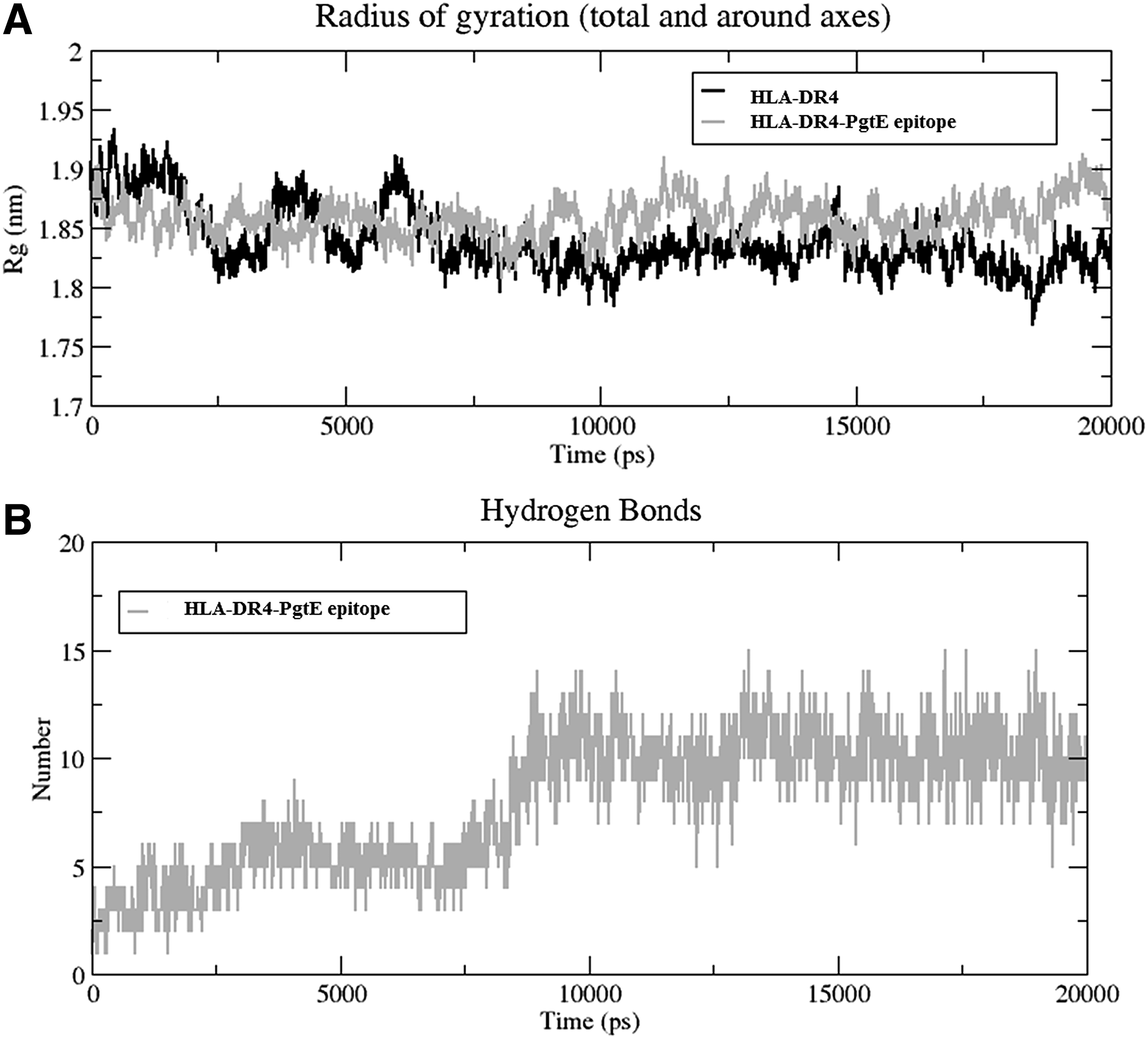
The analysis confirms that PgtE epitope interaction with HLA-DR4 can inhibit its functional activities. In addition, the increase in hydrogen bond pattern with HLA by the PgtE epitope interaction conveys that binding was more favored with a fourfold increase from the initial hydrogen bond numbers (Fig. 9) and it stabilized more than half of the time period of MD simulation (Fig. 8B). Overall, MD analysis reveals that PgtE epitope was binding and interacting firmly with the HLA, and hence, we are proposing that PgtE epitope would be a prominent epitope vaccine for the human pathogen, S. typhi, requiring further steps to be elevated as a vaccine drug in the near future.
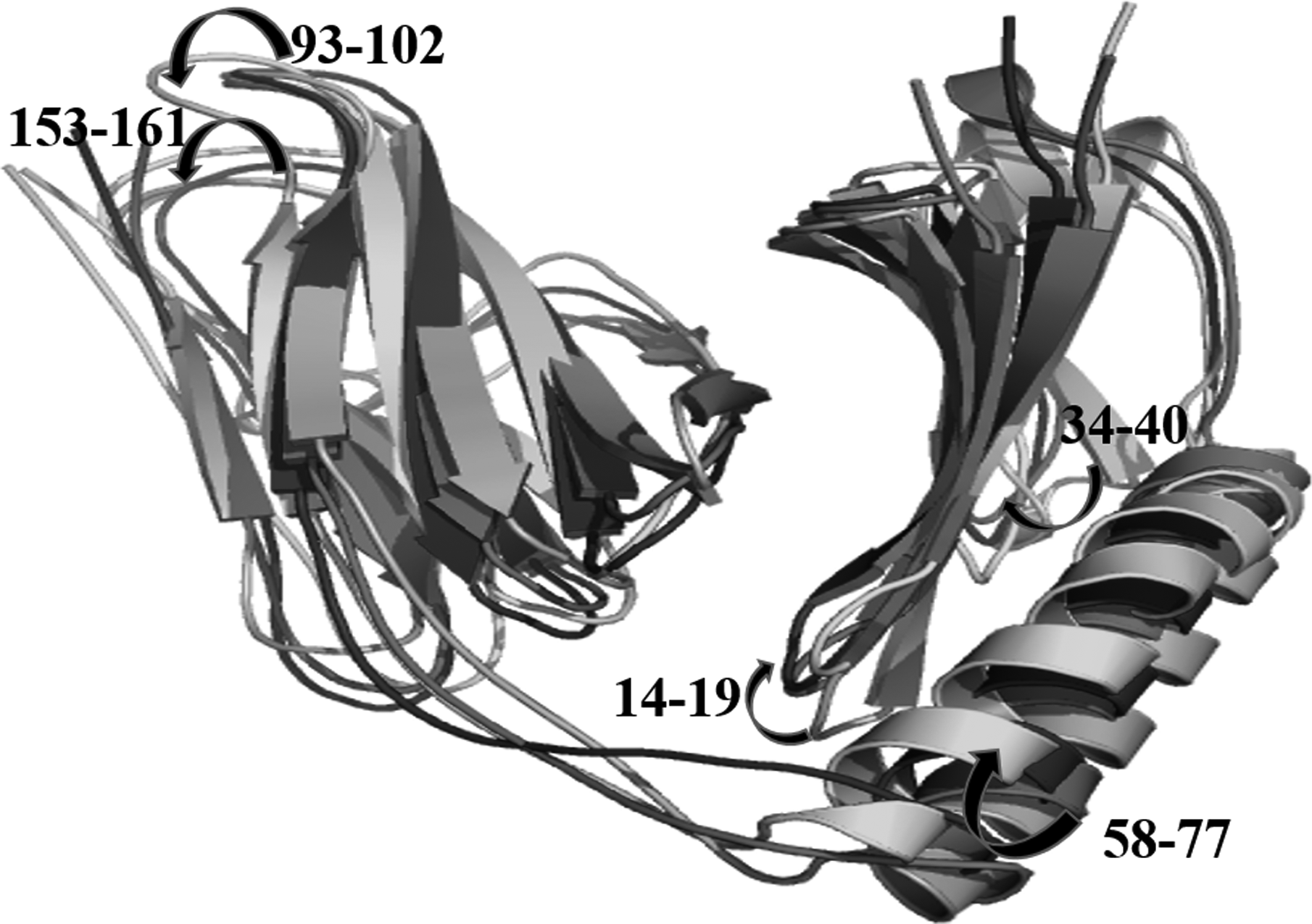
The large structural changes occurred in HLA, while the transformation processed from grey (initial) to light grey (12,770 ps) to dark grey (14,320 ps) to black (15,100 ps), where 14–19, 34–40, 58–77 (large helix), 93–102, 153–161 aa regions responded for flexibility.
4. Conclusion
The systematic identification of the PgtE epitopes (B cell and T cell) by computational-based analysis provided in-depth information on the possible function of S. typhi in typhoid pathogenesis. According to molecular interaction studies, PgtE (HIS180, TYR187, and ILE 179) was in frequent contact with HLA-DR4 and this complex structure was more stable throughout the molecular dynamics simulation (20 ns). In addition to that, the detailed interaction between the generated PgtE (179 IHPDTSANY 187) epitopes with the HLA-DR4 model allows a hypothetical analysis of potential therapeutic strategy for typhoid infections. The identified epitopes also require proper experimental validation as vaccine against typhoid fever.
Footnotes
Acknowledgment
The authors thank Dr. M. Beutline Malgija, Department of Bioinformatics, Bharathiar University, Coimbatore, Tamil Nadu, India, for valuable suggestions to the article.
Author Disclosure Statement
The authors declare that there are no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.