
Abstract
Abstract
The alignment among three or more nucleotides/amino acids sequences at the same time is known as multiple sequence alignment (MSA), a nondeterministic polynomial time (NP)-hard optimization problem. The time complexity of finding an optimal alignment raises exponentially when the number of sequences to align increases. In this work, we deal with a multiobjective version of the MSA problem wherein the goal is to simultaneously optimize the accuracy and conservation of the alignment. A parallel version of the hybrid multiobjective memetic metaheuristics for MSA is proposed. To evaluate the parallel performance of our proposal, we have selected a pull of data sets with different number of sequences (up to 1000 sequences) and study its parallel performance against other well-known parallel metaheuristics published in the literature, such as MSAProbs, tree-based consistency objective function for alignment evaluation (T-Coffee), Clustal
1. Background
M
The MSA problem is a nondeterministic polynomial time (NP)-complete optimization problem (Dogan and Otu, 2014); therefore, different heuristic approaches have been developed for solving this problem in an efficient amount of time. In the literature, we find three main groups: progressive methods, consistency-based methods, and iterative refinement methods.
The most representative progressive methods are Clustal W (Thompson et al., 1994), Clustal
The second group includes those methods based on consistency. These approaches construct a database of local and global alignments between each pair of sequences that helps to build an accurate multiple alignment among all the given sequences. Among the most important consistency-based tools are tree-based consistency objective function for alignment evaluation (T-Coffee) (Notredame et al., 2000), PROBabilistic CONSistency-based MSA (ProbCons) (Do et al., 2005), and MSAProbs (Liu et al., 2010).
Finally, we find the iterative refinement tools. The methodology followed by these tools starts by performing a progressive alignment, and then they iterate with the aim of correcting any possible inaccurate gap inserted in the progressive construction stage. The refinement process is repeated until no further improvements are found or until a predefined number of iterations are reached. Among the most widely used iterative refinement methods, we find multiple sequence comparison by log-expectation (Edgar, 2004) and multiple alignment using fast Fourier transform (MAFFT) (Katoh et al., 2002).
Multiobjective optimization in conjunction with evolutionary computation is a powerful methodology to optimize real-world problems in the bioinformatics (Gonzalez-Alvarez et al., 2015) and telecommunication fields (Rubio-Largo et al., 2013a; Rubio-Largo et al., 2013b). In Rubio-Largo et al. (2016), we propose a hybrid multiobjective memetic metaheuristic for the multiple sequence alignment (H4MSA) for optimizing the quality and consistency of the final alignment simultaneously. The results reveal that H4MSA is a very promising tool.
Given a set of k nonaligned sequences wherein the length of the largest sequence is L, the time and space complexity for solving the MSA problem is O(k2k Lk) (Waterman et al., 1976); therefore, finding an optimal alignment of a large number of sequences (
The main disadvantage of H4MSA is its prohibitive running time when the number of sequences increases. The main contribution of this article is an efficient parallel version of H4MSA for aligning sets of sequences with a large number of sequences accurately in a reasonable amount of time.
1.1. MSA problem
Given a set of sequences S:
An MSA of S is defined as
In this way, a multiple alignment is obtained by adding gaps to the sequences of S so that their lengths become the same. It can be seen as a matrix representation where the rows represent sequences and the columns represent aligned symbols. Each column of an alignment must contain at least one symbol of • Unaligned sequences (input): s1: TSREETKKKHPDASVNFSEFSKKCSERWKTMSAKEKGKFEDMA (43) s2: RKTALENPRMRNSEISKQLGYQWKMLTEAEKWPFFQEAQKLQAMH (45) s3: FFMEKTAKYAKLHPEMSNSDLTKILSKKWKELPEKKKMKYIQDFQREKQEF (51) s4: VVAESTLKESSAINKILGRRWHALSREEQAKYYELARKERQLHMQL (46) s5: DYKKETESDIDKHLSDIWTVNKGSWVALGFSDGQEA (36) • Aligned sequences (output):] * * * *
To find an accurate alignment, we propose the use of multiobjective optimization. Therefore, we search the best solution (alignment) that simultaneously maximizes the weighted sum-of-pairs (WSPs) function with affine gap penalties (f1) (Gupta et al., 1995) and the number of totally conserved (f2) columns score (Edgar, 2004; Thompson et al., 2005).
On the one hand, the WSPs with affine gaps (f1) need to maximize the following equations:
In Equation 1, AL is the alignment length, SP(l) is the sum-of-pairs score of the
On the other hand, the number of totally conserved (f2) columns score refers to the number of columns that are completely aligned with exactly the same compound. This objective function needs to be maximized to ensure more conserved regions within the alignment. In the aforementioned example, the symbol “*” indicates a column completely aligned with exactly the same compound; therefore, f2 = 4.
The maximum number of columns (alignment length) was limited to
The rest of the article is organized as follows. A detailed description of the parallel H4MSA algorithm is presented in Section 2. Section 3 is devoted to compare the alignment accuracy and parallel efficiency of H4MSA with other parallel approaches published in the literature. Section 4 discusses the obtained results, whereas Section 5 concludes the article and suggests future research avenues.
2. Methods
This section is divided into two parts. The first one is devoted to describe the H4MSA algorithm and study the points in which the algorithm spends more time. The second part presents a detailed description about the parallelization process of the metaheuristics.
2.1. H4MSA
The shuffled frog-leaping optimization algorithm (SFLA) is a memetic metaheuristic developed by Eusuff et al. (2006). The search procedure begins with a random population of frogs (solutions) in a swamp.
The population of frogs is divided into isolated communities (memeplexes) that will evolve independently, allowing different directions within the search space. At each community, the frogs evolve by sharing their ideas with their neighbor frogs. In this way, those frogs with better ideas will share more ideas than those frogs with poor ideas.
In addition, the best frogs of each community will share their ideas with other frogs in different communities. After a number of iterations, the communities of frogs are forced to mix and new communities are formed through a shuffling process to accelerate the convergence of the algorithm.
The chromosome representation of a solution in H4MSA is different from the traditional binary representation (1 indicates gap symbol and 0 indicates a residue). For example, the binary representation of the alignment shown in Section 1.1 is:
However, in H4MSA, a solution only stores the number of groups of gaps followed by the information of each group: position of the first gap and number of successive gaps (negative value). The chromosome representation of the example alignment will be
Given a set of k unaligned sequences (S), m memeplexes (number of communities) with n frogs per memeplex, a fixed number of evolutionary steps (N), and a stopping criterion, the procedure of H4MSA is as follows:
1. Generate and evaluate 2. Sort the 3. Divide the 4. For each memeplex (Yi) (a) Select the local worst ( (b)
and the following portion of the local worst (
and the resultant new frog (
(c) Apply the following mutation process to (i) Move a block: randomly select a block of gaps/compounds and move it one position toward the left or right.
(ii) Merge two groups: randomly select one of the sequences, choose a group of gaps/compounds, and merge it with the closest group.
(iii) Divide a group: randomly select one of the sequences, choose a group of gaps/compounds, and divide it into two new groups of approximately the same size.
(iv) Compact alignment: delete those columns with all gaps.
(d) Evaluate (e) Select the global best frog ( (f) (g) Apply the mutation process to (h) Evaluate (i) Apply a Local Search to
(j) Replace (k) If the maximum number of evolutionary steps (N) has not been reached, then go to Step 4(a). Otherwise, continue with the next memeplex. 5. Merge the frogs from the m memeplexes. 6. If the stopping criterion is satisfied, output the set of nondominated solutions; otherwise, go to Step 2.








As we can see, the output of H4MSA is a set of nondominated solutions, that is, a set of solutions that represents a trade-off between alignment quality (f1) and consistency (f2).
2.2. Parallel scheme
In this work, we propose a parallel scheme of H4MSA for a shared memory architecture. After studying the computational requirements of H4MSA, we can see that a large amount of time is spent in the initial generation of the population and in the evolving process of each memeplex; therefore, we focus on parallelizing these tasks. In Figure 1, we present a flowchart of the parallel scheme of H4MSA.
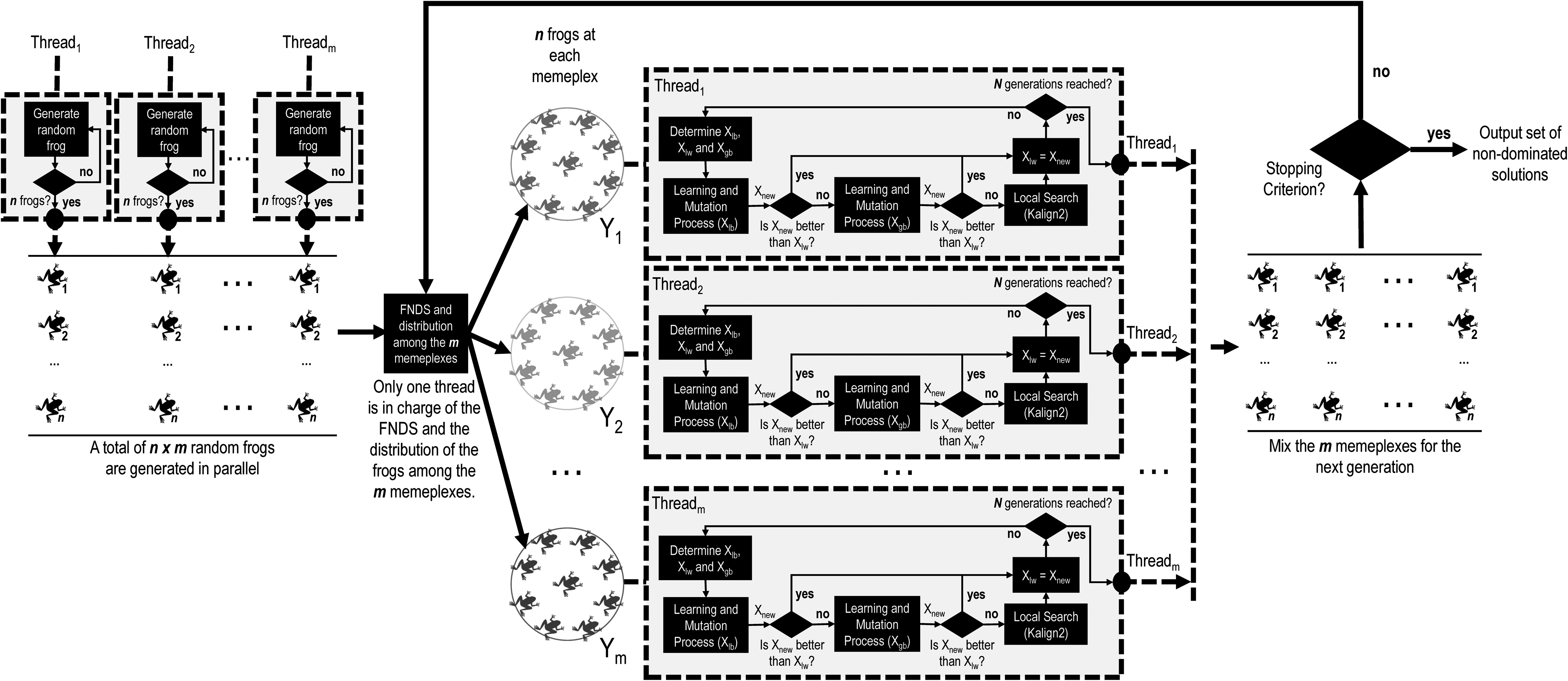
Parallel flowchart of H4MSA. FNDS, fast non-dominated sorting; H4MSA, hybrid multiobjective memetic metaheuristic for the multiple sequence alignment.
As we can see, in the generation of the initial population (Step 1), the
The second and third steps of H4MSA (Fast Nondominated Sorting procedure and the shuffle procedure) are carried out in a single thread mode, that is to say, only one thread is in charge of carrying out these two steps. The main reason is because the sorting and division process consumes no significant runtime in comparison with the other tasks of H4MSA. These steps consume ∼0.1% of a single generation runtime.
In Step 4, H4MSA performs m independent evolutionary processes of N iterations. It is clear that the tasks carried out within each evolutionary process (learning process, mutation process, and local search) are computationally expensive. Therefore, each of the m threads is in charge of evolving a memeplex in parallel. The workload of each memeplex may be different for each thread; therefore, instead of using a static distribution where each of the m threads is in charge of performing the consecutive
Since H4MSA has been implemented in OpenMP, we have parallelized the evolving of each memeplex by using the dynamic schedule. This schedule uses an internal work queue with the loop iterations to each thread. When a thread is finished, it retrieves the next loop iteration from the top of the work queue. The number of iterations performed by each thread is decided during the execution of the algorithm; so, threads may do different number of iterations. In Figure 2, we show the advantages of using a dynamic distribution of the iterations among threads when the workload of iterations is different. As we can see, 51.51% of efficiency is achieved with a static schedule; however, the efficiency increases up to 94.45% when the distribution of the iterations is dynamic.
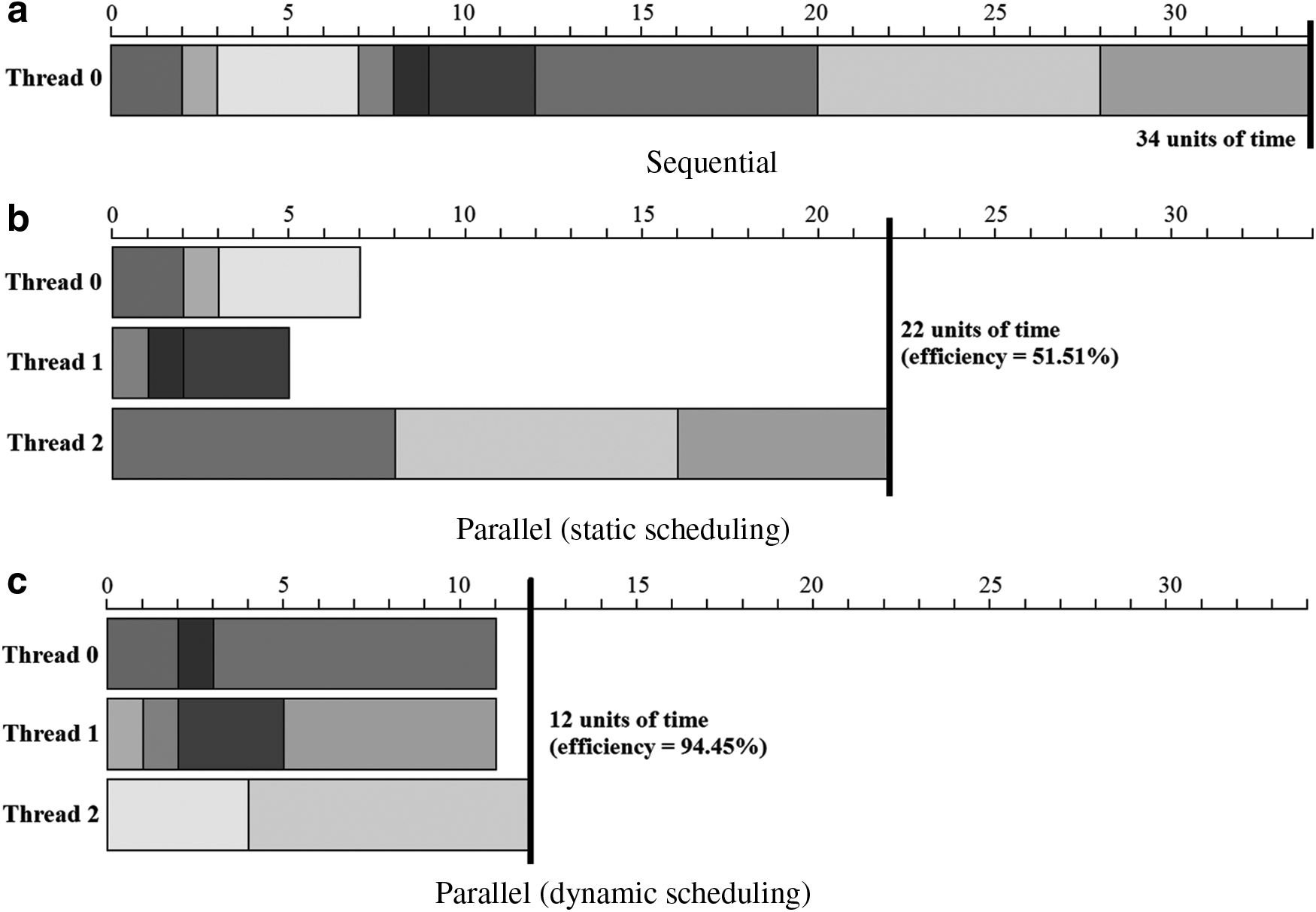
Examples of sequential execution (
3. Results
In this section, we present a comparative study between our parallel approach (H4MSA) and other multithreaded MSA approaches published in the literature.
We have compared the multithreaded version of H4MSA with those MSA approaches that allow being run in multicore environments:
• MSAProbs (version 0.9.7) (Liu et al., 2010) It is a parallel and accurate approach for MSA. It allows the use of the -num_threads flag to specify the number of threads. • T-Coffee (version 11.00.8) (Notredame et al., 2000). It is a widely used MSA approach in the field. The steps of T-Coffee are multithreaded by using the -multi_core flag, specifying the number of cores to use by the -n_core flag. • Clustal • MAFFT (version 7.215) (Katoh et al., 2002). It is a method for rapid MSA based on fast Fourier transform. From version 6.8, MAFFT switches to the multicore version by simply specifying the number of threads with the—thread flag.
The aforementioned approaches were run by using the default parameter configuration.
In H4MSA, we found three main parameters: number of memeplexes (m), number of frogs at each memeplex (n), and the number of evolutionary steps (N). In this comparative study, we have used m = M (number of memeplexes equal to number of threads), n = 32 (32 frogs per memeplex), and N = 10 (10 evolutionary steps). The stopping criterion is based on the number of fitness evaluations: 50,000 evaluations.
The data sets used in these experiments were taken from the HOMFAM benchmark suite (Sievers et al., 2011). As we can see in Table 1, we have chosen data sets with different number of sequences, from 88 sequences to 1056 sequences, to evaluate the performance of the approaches when the number of sequences increases.
To extract useful conclusions with a certain level of statistical confidence, 30 independent runs were performed for each approach involved, and the average runtime was used. The architecture selected for conducting these experiments was a cluster with two AMD Opteron™ Processor 6376 of 16 cores (a total of 32 cores) at 2.3 GHz, 12 MB Cache L3, 48 GB of DDR3 RAM, and Scientific Linux 6.1.
For measuring the performance of the parallel approaches, we have used two well-known metrics: speedup and efficiency. Amdahl's Law states that the performance improvement to be gained from using some faster mode of execution is limited by the fraction of the time the faster mode can be used. Therefore, Amdahl's Law defines the speedup that can be gained by using a particular feature. In a more formal way, let TM be the runtime for an algorithm using M threads and T1 the runtime of the sequential version, the speedup reports us how much faster an algorithm will run as opposed to the sequential version. The efficiency is computed by dividing the obtained speedup with M threads by the number of threads used (M).
To determine the accuracy of each MSA method, we have evaluated the level of conservation with the BLOSUM62 substitution matrix. Therefore, the alignment obtained by each approach for each data set was scored by using the +evaluate blosum62mt action provided by T-Coffee. Note that a higher score implies better alignment accuracy.
4. Discussion
This section discusses the obtained results and allows to draw important considerations with respect to the proposed system.
On one hand, in Table 2, we present the conservation score obtained by H4MSA, MSAProbs, T-Coffee, Clustal
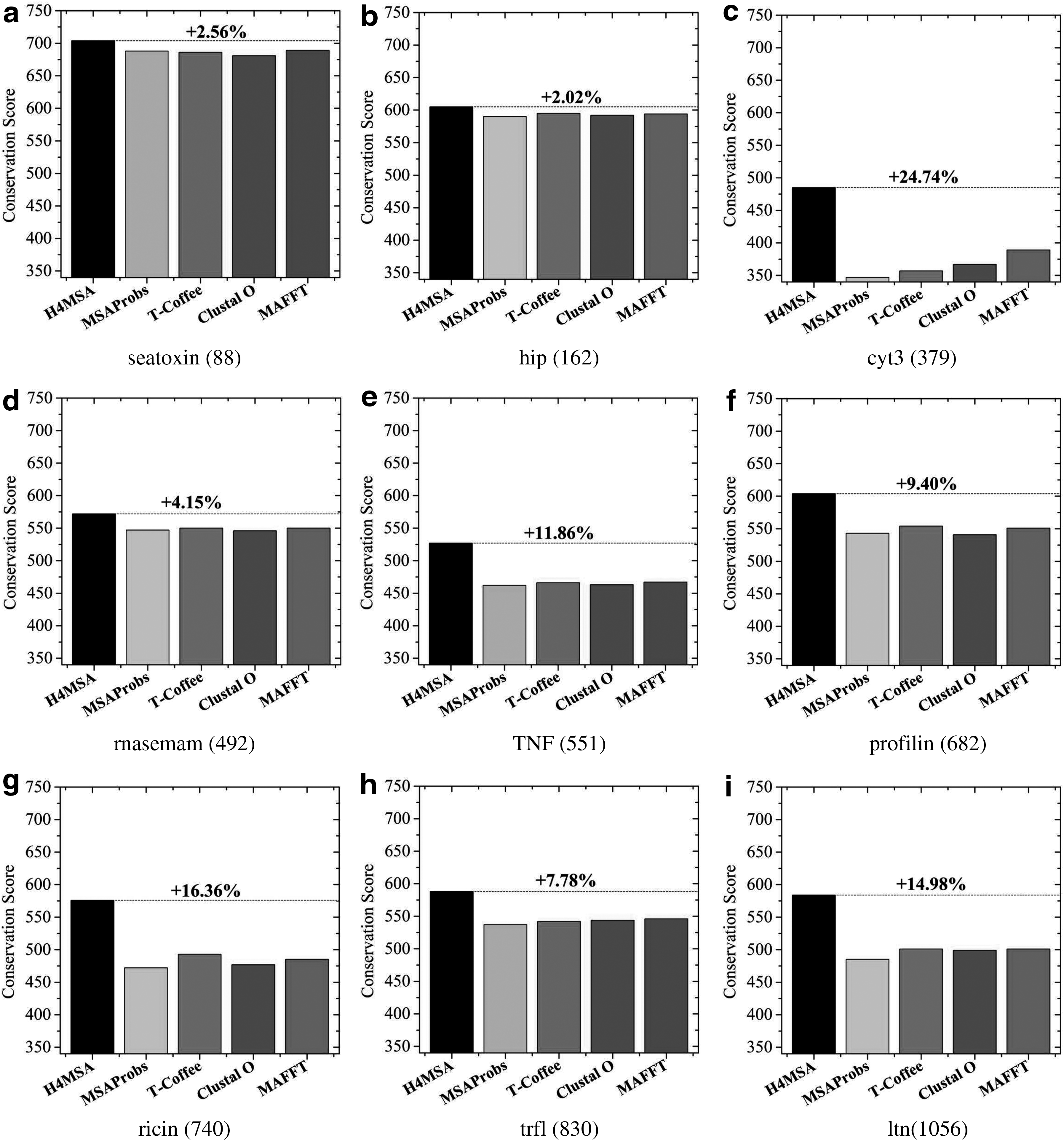
Comparison among H4MSA, MSAProbs, T-Coffee, Clustal
H4MSA, hybrid multiobjective memetic metaheuristic for the multiple sequence alignment; MAFFT, multiple alignment using fast Fourier transform; MSA, multiple sequence alignment; T-Coffee, tree-based consistency objective function for alignment evaluation.
Bold represents the aligner with the highest average conservation score.
To ensure that the differences among the approaches in our comparative study are statistically significant, we perform a Wilcoxon signed-rank test between each pair of methods by using a confidence level of 1% (p-value <0.01). As we can see in Table 3, we can conclude that the differences of conservation score between H4MSA and any other method result statistically significant in the nine benchmarks tested.
On the other hand, we compare the parallel performance of the approaches under study. In Table 4, we present the parallel speedup and efficiency obtained with different number of cores (M = 2, 4, 8, 16, and 32 cores). As we can see, the parallel performance of Clustal
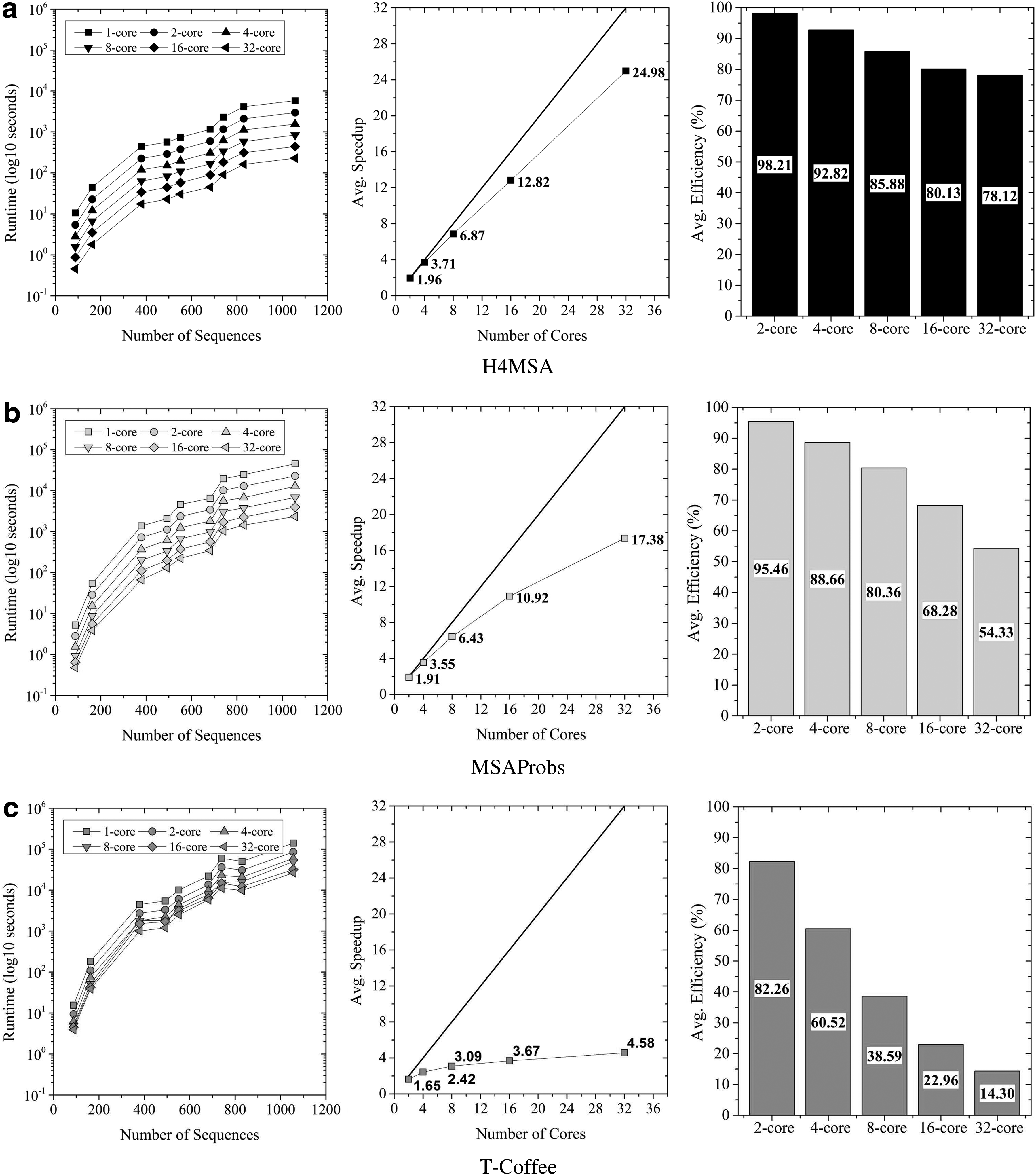
Runtime, average speedup, and average efficiency (%) obtained by each MSA tool. H4MSA (
S: Average speedup in the nine data sets of HOMFAM.
E: Average efficiency (%) in the nine data sets of HOMFAM.
In Figure 4, if we compare the sequential runtime of each approach, we can see that the fastest algorithms are (in order): Clustal
All in all, we can conclude that H4MSA is not only an accurate alignment method but also its parallel performance allows it to handle data sets with hundreds of sequences in a reasonable amount of time.
5. Conclusions
A parallelization of the H4MSA is presented in this work. H4MSA is based on the SFLA, which provides the benefits of information mixture of the “shuffled complex evolution” technique. The parallel version of H4MSA has been compared with the parallel approaches of MSAProbs, T-Coffee, Clustal
As future work, with the aim of solving larger data sets, we intend to develop a parallel version of H4MSA for shared- and distributed-memory architectures. In addition, we intend to develop other parallel swarm intelligence metaheuristics, for example, Rubio-Largo et al.(2012).
Footnotes
Acknowledgments
This work was partially funded by the AEI (State Research Agency, Spain) and the ERDF (European Regional Development Fund, EU), under the contract TIN2016-76259-P (PROTEIN project). Álvaro Rubio-Largo is supported by the postdoctoral fellowship SFRH/BPD/100872/2014 granted by Fundação para a Ciência e a Tecnologia (FCT), Portugal. Mauro Castelli and Leonardo Vanneschi are supported by project PERSEIDS (PTDC/EMS-SIS/0642/2014) and BiolSI RD unit (UID/MULTI/04046/2013), funded by FCT/MCTES/PIDDAC, Portugal.
Author Disclosure Statement
No competing financial interests exist.