
Abstract
Abstract
Estimation of heritability is an important task in genetics. The use of linear mixed models (LMMs) to determine narrow-sense single-nucleotide polymorphism (SNP)-heritability and related quantities has received much recent attention, due of its ability to account for variants with small effect sizes. Typically, heritability estimation under LMMs uses the restricted maximum likelihood (REML) approach. The common way to report the uncertainty in REML estimation uses standard errors (SEs), which rely on asymptotic properties. However, these assumptions are often violated because of the bounded parameter space, statistical dependencies, and limited sample size, leading to biased estimates and inflated or deflated confidence intervals (CIs). In addition, for larger data sets (e.g., tens of thousands of individuals), the construction of SEs itself may require considerable time, as it requires expensive matrix inversions and multiplications. Here, we present FIESTA (Fast confidence IntErvals using STochastic Approximation), a method for constructing accurate CIs. FIESTA is based on parametric bootstrap sampling, and, therefore, avoids unjustified assumptions on the distribution of the heritability estimator. FIESTA uses stochastic approximation techniques, which accelerate the construction of CIs by several orders of magnitude, compared with previous approaches as well as to the analytical approximation used by SEs. FIESTA builds accurate CIs rapidly, for example, requiring only several seconds for data sets of tens of thousands of individuals, making FIESTA a very fast solution to the problem of building accurate CIs for heritability for all data set sizes.
1. Introduction
H
To cope with this challenge, linear mixed model (LMM) approaches (Kang et al., 2008, 2010; Visscher et al., 2008; Lippert et al., 2011; Vattikuti et al., 2012; Zhou and Stephens, 2012; Wright et al., 2014; Kruijer et al., 2015) have been applied to estimate the heritability explained by common SNPs (the narrow-sense SNP-heritability, to which we refer as heritability, and denote by h2) from cohorts of unrelated individuals, such as those found in GWASs (Yang et al., 2010). Estimation under the LMM is usually performed using restricted maximum likelihood (REML) estimation, and is implemented in some widely used tools, like the Genome-wide Complex Trait Analysis (GCTA) software package (Yang et al., 2011). LMMs utilize all variants from a GWAS and not just the variants that are statistically significant and, therefore, are able to account for variants with small effect sizes.
As in any statistical analysis, the process of estimating the heritability suffers from statistical uncertainty. Typically, confidence intervals (CIs) are reported alongside with point estimates to quantify this uncertainty. Usually, such CIs are constructed from standard errors (SEs), which make the assumption that the estimators asymptotically follow a normal distribution. However, it has been shown (Lohr and Divan, 1997; Burch, 2007; Burch, 2011; Kruijer et al., 2015; Schweiger et al., 2016) that such CIs can be highly inaccurate. This is because estimators do not necessarily obey the conditions required for them to asymptotically follow the normal distribution. In addition, these CIs may spread beyond the natural boundaries of their parameters, for example, including negative values for heritability. As a result, these CIs are often inaccurate, difficult to interpret, or lead to erroneous conclusions.
To handle these issues, previous approaches have taken several directions. Nonstandard asymptotic theory for boundary and near-boundary maximum likelihood estimates has been developed (e.g., Chernoff, 1954; Moran, 1971; Self and Liang, 1987), and it has been suggested to replace the asymptotic normality assumption with the asymptotics developed for the nonstandard boundary case (Stern and Welsh, 2000). Visscher and Goddard, (2015) derived an analytical expression for the asymptotic variance of the heritability estimator in a range of pedigree- and marker-based experimental designs. Unfortunately, these conditions typically do not hold for genomic data sets, mainly due to the limited sample size, making either of these approximations ineffective (Schweiger et al., 2016). Other approaches include hierarchical bootstrapping schemes (e.g., Thai et al., 2013), extending the REML estimation method with Bayesian priors (e.g., Wolfinger and Kass, 2000; Chung et al., 2011), using alternative statistics as a basis for building CIs (Harville and Fenech, 1985; Burch and Iyer, 1997; Burch, 2007), or using Bayesian posterior distribution of the heritability value (Furlotte et al., 2014).
An alternative approach is the parametric bootstrap test inversion technique, which constructs CIs through sampling phenotypes, performing heritability estimation on the sampled phenotypes, estimating the distribution of the heritability estimator, and using these estimates as a basis for CI construction (Carpenter and Bithell, 2000). The main advantage of using a parametric bootstrap approach is that it does not require any assumptions on the distribution of the heritability estimator or of Bayesian priors. As a naive implementation of this approach would be computationally prohibitive, the Accurate LMM-based heritability Bootstrap confidence Intervals (ALBI) method (Schweiger et al., 2016) utilizes a highly accurate approximation that allows an efficient construction of accurate CIs. However, ALBI still requires a preprocessing step. Newer data sets [e.g., the UK Biobank (Sudlow et al., 2015)] may contain tens or hundreds of thousands of individuals, for which this step may require hours of computation time. In addition, the need for a preprocessing step can be an obstacle in the adoption of a better CI construction method.
In this article, we introduce FIESTA (Fast confidence IntErvals using STochastic Approximation), which dramatically reduces the running time of CI construction by several orders of magnitude, for example, to mere seconds for data set with tens of thousands of individuals, compared with hours or days. The key ingredient of our approach is a CI construction algorithm from the field of stochastic approximation (for a review, see Kushner and Yin, 2003). Originating in the work of Robbins and Monro (1951), stochastic approximation algorithms are recursive update rules that can be used, among other things, to solve optimization problems or function inversion problems when the collected data are subject to noise. It has been shown (Garthwaite and Buckland, 1992) that stochastic approximation can be used to construct CIs for general families of parametric distributions, given the ability to randomly sample from them, and this is the approach we employ here. We validate FIESTA on two real data sets, the Northern Finland Birth Cohort (NFBC) data set (Sabatti et al., 2009) and the Wellcome Trust Case Control Consortium 2 (WTCCC2) (Sawcer et al., 2011) data set.
In addition to the significant speedup in time, FIESTA requires no preprocessing step beyond calculating the eigendecomposition of the kinship matrix, which is usually already performed as a part of heritability estimation. Finally, we show that FIESTA is even significantly faster than the analytical SE formulation. In summary, FIESTA can effectively be used extremely easily to rapidly generate accurate CIs for REML heritability estimates. FIESTA is available as part of the ALBI toolkit at https://github.com/cozygene/albi
2. Results
2.1. A faster method for calculating CIs for heritability
CIs constructed from SEs, which are based on the assumption of a normal distribution for the heritability estimators, were previously shown to be inaccurate (Lohr and Divan, 1997; Burch, 2007; Burch, 2011; Kraemer, 2012; Kruijer et al., 2015; Schweiger et al., 2016). In this article, we introduce FIESTA, a method that generates accurate CIs for h2, the true heritability value, given
The methodology of test inversion can be described as follows. The estimator
It remains to define suitable acceptance regions. In section 3, we review our scheme for defining acceptance regions. A basic ingredient of our construction of acceptance regions is inverting certain quantile functions of the distribution of
Instead of carrying out this task by a full parametric bootstrap estimate of the distribution of the estimator, we employ a technique from the field of stochastic approximation to achieve the same results with a fraction of the computational cost. The modified Robbins–Monro procedure (Joseph, 2004), described in section 3, is an iterative method that finds the inverse of the quantile function of a one-parameter distribution. It operates by iteratively (1) drawing a sample with a true heritability value equal to our current guess for the required inverse value, (2) comparing its estimated heritability to H2, (3) updating our current guess accordingly, by moving in the right direction, with a step size that decreases with the number of iterations. An additional speedup is acquired by using a fast method to calculate the derivative of likelihood of the sample, and using the derivative to compare its estimated heritability to H2, instead of performing the full likelihood maximization.
We applied FIESTA to construct 95% CIs for the NFBC data set (Sabatti et al., 2009) and the WTCCC2 data set (Sawcer et al., 2011), as seen in Figure 1. We then turned to verify the accuracy of these CIs, which can be measured as follows. Draw multiple phenotype vectors from the distribution assumed by the LMM with parameters that correspond to a true heritability value h2. From each such phenotype, construct a CI for its estimated heritability with a confidence level of, for example, 95%. If the constructed CIs are accurate, then they should cover the true underlying h2 95% of the time. Then, check the percentage of times in which the CI covered h2, as a function of h2. We measured the accuracy of FIESTA, with CIs designed to have a coverage of 95%. The results are shown in Figure 2, demonstrating that FIESTA accurately achieves the desired confidence levels.
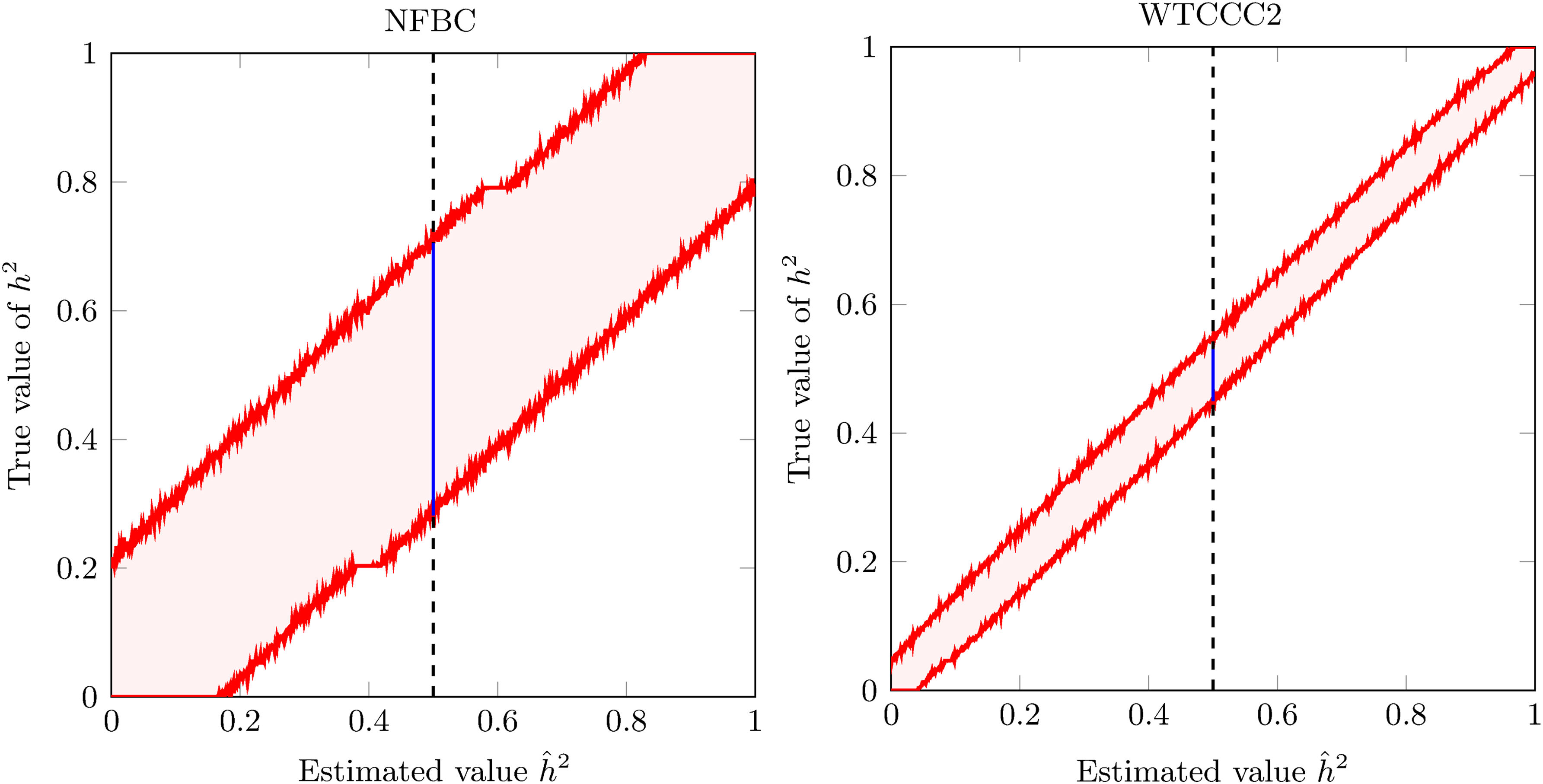
The 95% CIs for the NFBC and WTCCC2 data sets. Accurate 95% CIs constructed for the NFBC data set (Sabatti et al., 2009) (left) and the WTCCC2 (Sawcer et al., 2011) data set (right) by FIESTA. For each

Accuracy of CIs for the NFBC and WTCCC2 data sets. The coverage probabilities of the FIESTA CIs. The coverage probabilities are shown for CIs designed to have coverage probabilities of 95%. The CIs achieve accurate coverage.
2.2. Benchmarks
We compared the speed of the stochastic approximation approach, implemented in FIESTA, with that of using the parametric bootstrap for estimating the distribution of heritability estimator. The latter was tested either as implemented naively by using either GCTA (Yang et al., 2011) and pylmm (Furlotte and Eskin, 2015) or by using ALBI (Schweiger et al., 2016). Both the stochastic approximation and parametric bootstrap approaches require the calculation of the eigendecomposition of the kinship matrix. As this is already often a part of the heritability estimation algorithm, its calculation time is separated in the benchmarks. In section 4, we discuss how this step could be avoided altogether.
One difference between the two approaches is that the bootstrap approach performs a lengthy preprocessing step that estimates many distributions. Once these distributions are estimated, constructing a CI is very rapid. In contrast, the stochastic approximation approach does not perform a preprocessing step, but performs a nontrivial calculation per CI.
The construction of a single CI with FIESTA consists of calculating six to eight values using the modified Robbins–Monro procedure (see section 3). The first four values depend only on the kinship matrix, but not on the heritability estimate for which we construct a CI, so they need to be calculated only once per kinship matrix, and can then be shared between several CIs. Each modified Robbins–Monro run has the complexity of
We also compared FIESTA with the performance of the analytical SE approach. Although often inaccurate, analytical SEs are often the go-to method by many practitioners: first, their calculation is conceptually easy to understand, since a closed-form formula exists for the SEs (see section 5); second, using a closed-form expression is often perceived as faster than more involved algorithmic procedures. However, this is not the case for heritability estimation, as SEs are calculated using variants of the Fisher information matrix [e.g., the average information (AI) matrix, as in GCTA (Yang et al., 2011)], whose calculation requires matrix-by-vector multiplications, which are
We performed a benchmark to evaluate FIESTA, using the NFBC and WTCCC2 data sets. We estimated the distributions of
Running times of FIESTA, compared with previous methods (see section 2 for more details). Running times are reported for the NFBC (2520 individuals) and WTCCC2 (13,950 individuals) data sets.
ALBI, Accurate LMM-based heritability Bootstrap confidence Intervals; CI, confidence interval; FIESTA, Fast confidence IntErvals using STochastic Approximation; GCTA, Genome-wide Complex Trait Analysis; n/a, not applicable; NFBC, Northern Finland Birth Cohort; SEs, standard errors; WTCCC2, Wellcome Trust Case Control Consortium 2.
As a comparison, we used FIESTA to construct varying number of CIs, using 1000 iterations in the modified Robbins–Monro procedure (see section 3). In Table 1, it can be seen that FIESTA is significantly faster, particularly when few CIs are needed. We also note that FIESTA is currently implemented in the Python language, using the numpy package; a significant additional speedup can be obtained by migrating to a compiled language, for example, C++.
We then continued to investigate the stability of CI construction and its dependency on the number of iterations. We ran FIESTA 100 times to construct CI for the NFBC and WTCCC2 data sets using 200, 500, 1000, or 2000 iterations. We measured the variance in the constructed CI endpoints (Table 2). As expected, the variance decreases with the number of iterations. In addition, we measured the mean and variance of the coverage of CIs under a grid of true heritability values. Here, also, we observed that variance of coverage decreases with the number of iterations. We note that 500 iterations are sufficient for reasonably accurate CIs for these data sets, and that the coverage of even 200 iterations is only slightly biased downwards.
Ninety-five percent CIs for the NFBC and WTCCC2 data sets were constructed 100 times, with either 200, 500, 1000, or 2000 iterations. CIs were constructed for
3. Methods
For clarity of presentation, we begin by defining the heritability under the LMM, and briefly reviewing stochastic approximation and its relevance to finding CIs. Finally, we introduce FIESTA, our improved method for faster construction of CIs for heritability.
3.1. The LMM and REML
We consider the following standard LMM (see Searle et al., 2009 for a detailed review). Let n be the number of individuals and m be the number of SNPs. Let
Define
The narrow-sense heritability due to genotyped common SNPs is defined as the proportion of total variance explained by genetic factors (Visscher et al., 2008):
Defining
The most common way of estimating h2 is REML estimation. REML consists of maximizing the likelihood function associated with the projection of the phenotype onto the subspace orthongonal to that of the fixed effects of the model (Patterson and Thompson, 1971). In Schweiger et al. (2016), it is shown that the distribution of
3.2. CIs for h2
We wish to build CIs with a coverage probability of
Let
and
3.3. Using stochastic approximation to calculate CIs
Robbins–Monro
Stochastic approximation methods are a family of iterative stochastic optimization algorithms that attempt to find zeroes, inverses, or extrema of functions that cannot be computed directly, but only estimated through noisy observations. The classical Robbins–Monro algorithm presents a methodology for solving a function inversion problem, where the function is the expected value of a parametrized family of distributions. Namely, a function
where
Using Robbins–Monro to calculate CIs
Garthwaite and Buckland (1992) have used the Robbins–Monro process for finding the endpoints of CIs, as we now describe. We discuss the case of one-sided CIs, but the application to two-sided CIs is immediate.
Suppose that
If we define
In detail, denote by yn a random sample from the random variable
The procedure is shown to be fully asymptotic efficient if
The modified Robbins–Monro procedure
As already mentioned, if the optimal step size constant is known, this procedure is fully asymptotic efficient. However, it was empirically shown to work poorly for extreme quantiles. Joseph (2004) suggested a modification of this procedure, which is tuned to obtain optimal convergence speed. It uses the following update form:
Joseph allows the use of a different target value, Cn, in each iteration, instead of the required constant, C. The step sizes an and target values Cn are derived explicitly in Joseph (2004) to be optimal under a Bayesian analysis framework. As in Garthwaite and Buckland (1992), the optimal step size also uses
3.4. Using the modified Robbins–Monro procedure to obtain CIs for heritability
We now describe how to rapidly construct CIs for heritability. As already described, the first step is to find
As already detailed, we make repeated use of the following procedure: (1) draw a random sample
where
for
Similarly, for finding
The second step involves calculating the quantities
In practice, we used the following choices in the modified Robbins–Monro procedure: (1) we used
where z is the quantile function of the normal distribution and
3.5. The NFBC data set
We analyzed 5236 individuals from the NFBC data set, which consists of genotypes at 331,476 genotyped SNPs and 10 phenotypes (Sabatti et al., 2009). From each pair of individuals with relatedness of >0.025, one was reserved, resulting in 2520 individuals.
3.6. The WTCCC2 data set
We analyzed the WTCCC2 data set (Sawcer et al., 2011). In the multiple sclerosis and ulcerative colitis data sets, we used the same data processing described in Yang et al. (2014) to ensure consistency. In brief, U.K. controls and cases from both United Kingdom and non-United Kingdom were used. SNPs were removed with
4. Discussion
We have presented FIESTA, an efficient method for constructing accurate CIs using stochastic approximation. We have shown that FIESTA is very fast, while achieving exact coverage due to the fact that it does not rely on any assumptions of the distribution of the estimator. FIESTA is also faster than the analytical approximation used by SEs. Owing to its speed, FIESTA can be easily used for data sets with tens or hundreds of thousands of individuals.
FIESTA requires the eigendecomposition of the kinship matrix, whose computational complexity is cubic in the number of individuals. Although this is often a preliminary step in heritability estimation, it may be computationally prohibitive for larger data sets. Recent methods for heritability estimation (see Loh et al., 2015) utilize conjugate gradient methods to avoid cubic steps altogether. One direction of extension for FIESTA is devising a procedure to calculate the derivative of the restricted likelihood function using conjugate gradient methods, which are quadratic, but do not require the eigendecomposition.
We note that the CIs constructed by FIESTA are estimated under a set of assumptions, particularly that the data are generated from the LMM as described in the section 3. Deviations from these assumptions could result in inaccurate CIs. Specifically, we observed that when the genotype matrix is of low rank (e.g., in the case where duplicates are introduced), then the CIs calculated by FIESTA may be inaccurate. We, therefore, recommend removing duplicates and closely related individuals from the data before the application of FIESTA.
A common extension of the LMM is that of multiple variance components, where the genome is divided into distinct partitions (e.g., according to functional annotations or by chromosomes), and the relative genetic contribution of each partition is estimated instead. Another extension is that of multiple traits, where several phenotypes are estimated concurrently, allowing dependencies between them. In principle, the methodology behind FIESTA can be applied to the multiparametric case as well. However, there are several computational and conceptual hurdles that make this application highly nontrivial. First, a major difficulty rises from the fact that it is no longer necessarily possible to jointly diagonalize several kinship matrices. Thus, the computation of the derivatives of the logarithm of the restricted likelihood functions can no longer utilize the eigendecomposition. Second, the inversion of acceptance regions of multiple parameters results in confidence regions of more than one dimension. Although these have the required coverage probability, their shape may be difficult to report or to interpret easily (e.g., an ellipsoid). For example, hyper-rectangular confidence regions are often desirable (Sidak, 1967), as the marginal CI of each parameter has the same coverage probability as the confidence region. Therefore, multiparametric extensions remain a future direction of research.
5. Appendix
5.1. Variance of estimators
The main method of calculating the variance of the estimator, applied by all widely used linear mixed model methods, employs the Fisher information matrix, or a variant of which, possibly applying the delta method in addition Wasserman, L. (2013). All of statistics: A concise course in statistical inference. Springer Science & Business Media. The observed information matrix
GCTA uses the average information (Gilmour et al., 1995) matrix
where
Given the eigendecomposition of
5.2. CIs for heritability
Our approach is based on the duality between hypothesis testing and CIs. As the distribution of
Since the distribution of
Let
as described in Figure 3.
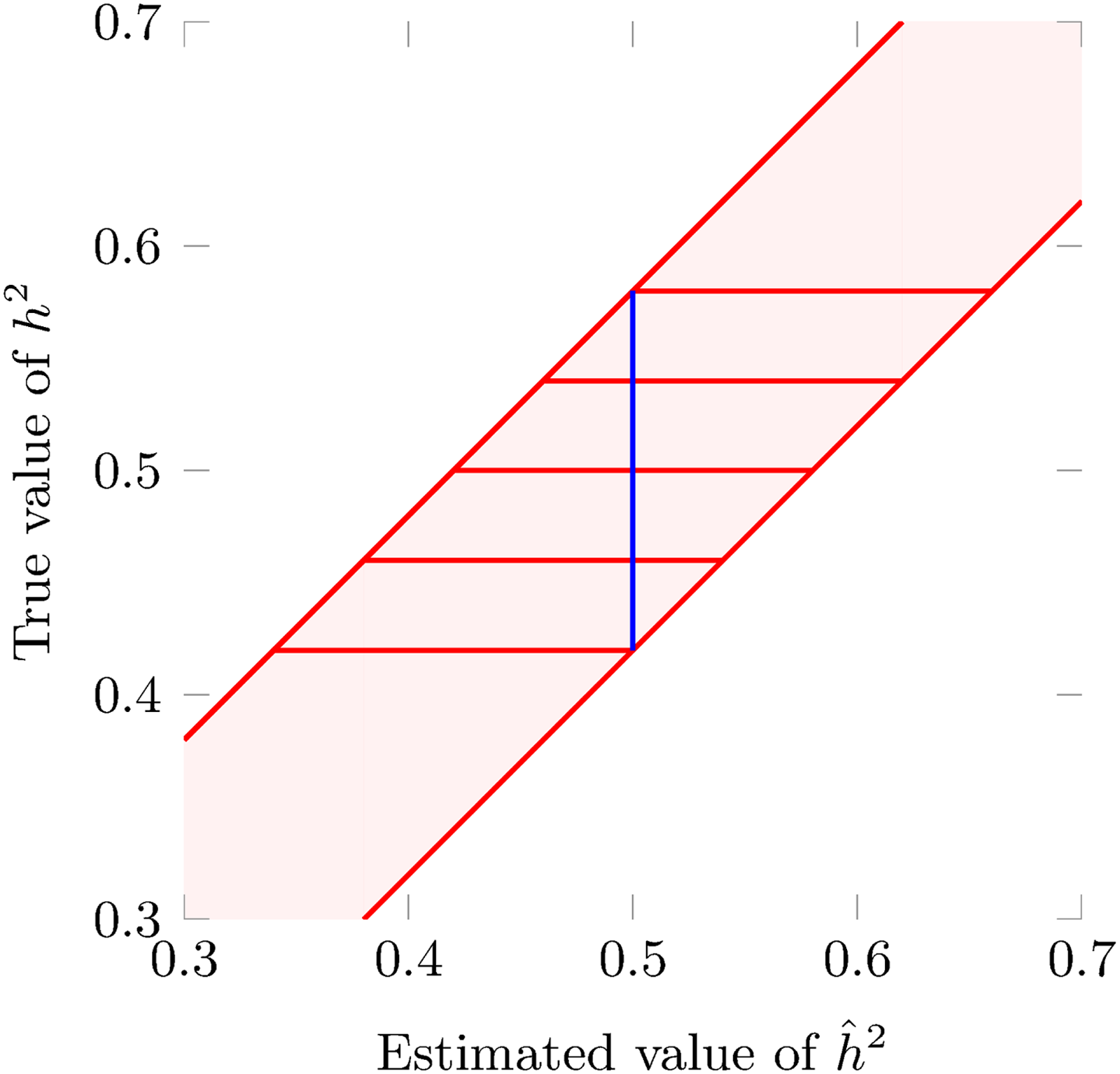
An illustration of acceptance regions and CIs. The diagonal lines are the
However, since the distribution is of a mixed type with discontinuity points, it may be the case that the probability of the interval
The following assumes s and t exist, and that
The three region types are illustrated in Figure 4. Inverting the acceptance regions, we get the following definition for
For the higher endpoint, we have

An illustration of the three acceptance region types. The diagonal lines, from left to right, indicate the quantile functions for
These conditions, phrased in terms of the quantile functions
and
It follows from the mentioned discussion that to construct a CI for an heritability estimate H2, we need to first find s, t as previously,
5.3. Accuracy of ALBI CIs

Accuracy of CIs for the NFBC and WTCCC2 data sets. The coverage probabilities of the ALBI CIs. The coverage probabilities are shown for CIs designed to have coverage probabilities of 95%.
Footnotes
Acknowledgments
The authors thank David Steinberg. R.S. is supported by the Colton Family Foundation. This study was supported, in part, by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University to R.S. The Northern Finland Birth Cohort data were obtained from dbGaP: phs000276.v2.p1. This study makes use of data generated by the Wellcome Trust Case Control Consortium. A full list of the investigators who contributed to the generation of the data is available in (![]() ). Funding for the project was provided by the Wellcome Trust under award 076113.
). Funding for the project was provided by the Wellcome Trust under award 076113.
Author Disclosure Statement
Regev Schweiger is an employee of MyHeritage Ltd.