
Abstract
Abstract
1. Introduction
1.1. Motivation
H
To reduce storage and transmission costs, raw sequencing data are often compressed using general purpose compression tools such as gzip based on Lempel-Ziv-77 (Ziv and Lempel, 1977) or bzip based on the Burrows-Wheeler Transform (Burrows and Wheeler, 1994). Although these classic tools compressed most of the public data, they are not optimized for HTS compression (Deorowicz and Grabowski, 2013; Holland and Lynch, 2013; Giancarlo et al., 2014; Hosseini et al., 2016; Numanagić et al., 2016). In FASTQ format, each record has three major components: (i) unique identifier, (ii) read sequence, and (iii) quality scores. A large variety of HTS-specific compression tools were proposed (Hach et al., 2012; Jones et al., 2012; Bonfield and Mahoney, 2013; Grabowski et al., 2014; Roguski and Deorowicz, 2014; Rozov et al., 2014; Saha and Rajasekaran, 2014; Benoit et al., 2015; Kingsford and Patro, 2015; Patro and Kingsford, 2015) to compress either FASTQ files or only the read sequences.
Although these tools are very efficient, they are not adapted to the context of large-scale sequencing projects that produce tens to several thousands of genomes per species. A pangenome, a set of genomes belonging to different strains of the same species, is characterized by a high degree of similarity and redundancy between the genomes (Tettelin et al., 2005). All HTS-specific compression tools can only consider redundancy and similarity within a single genome and not in a collection of genomes. Furthermore, large-scale sequencing projects such as the 1000 Genomes Project (1000 Genomes Project Consortium, 2015) may take years to complete, making the compression of continually growing pangenomes a challenging process.
1.2. Existing approaches
HTS-specific compression tools are divided into two categories: reference based and de novo. Reference-based methods generally provide high compression ratio by encoding similarities between the read sequences (reads) and a reference sequence (reference) usually by mapping the reads to the reference. These tools require that the reference used for compression is provided with the compressed archive for decompression, adding extra storage and transmission costs. Note that only a small fraction of sequenced species that are accessible in public databases have such a reference available. In contrast, de novo compression tools perform similarity search within a set of reads to exploit its redundancy. BARCODE (Rozov et al., 2014) is a reference-based method that makes use of cascading Bloom filters (BFs) (Salikhov et al., 2014) to compress reads. It inserts reads perfectly matching to a reference into a BF (Bloom, 1970) that can generate false positives. To reduce the number of false positives, BARCODE subsequently inserts them into cascading BFs to tell apart false positives from true positives. Kpath (Kingsford and Patro, 2015) constructs a de Bruijn graph (dBG) from the reference and encodes each read as a path within the graph.
The paths within the graph are then encoded through arithmetic coding (Witten et al., 1987). The beginnings of such paths are stored separately in a trie and encoded with LZ-77. QUIP (Jones et al., 2012) uses a lossless compression algorithm based on adaptive arithmetic encoding of the identifier, read, and quality score streams of the FASTQ format. A reference sequence and a sequence alignment of the reads can be used to improve compression of the reads. QUIP can also perform assembly-based compression. Similar methods are used in FASTQZ and FQZCOMP (Bonfield and Mahoney, 2013). SCALCE (Hach et al., 2012) uses core substrings as a measure of similarity to cluster similar reads together. Those core substrings are generated through Locally Consistent Parsing (Sahinalp and Vishkin, 1996). SCALCE compresses the reads in each cluster with gzip. ORCOM (Grabowski et al., 2014) reorders reads by similarity as well: it creates clusters of reads that share the same minimizer (Roberts et al., 2004), that is, the lexicographically smallest p-mer of each read with p usually between 8 and 15. Reads of the same cluster are then merged and compressed. Similar to ORCOM, Mince (Patro and Kingsford, 2015) uses the minimizer approach for clustering.
For each read to process, a set of candidate clusters is first established from the k-mers it is composed of. The read is then assigned to the candidate cluster that maximizes the number of q-mers they share. If the read has no candidate cluster, it is assigned to a new cluster corresponding to its minimizer of length k. DSRC 2 (Roguski and Deorowicz, 2014) compresses the different streams of FASTQ files with different methods: arithmetic coding, Huffman coding (Huffman, 1952), as well as 2 bits/base in the case of the DNA sequence stream. Finally, LEON (Benoit et al., 2015) encodes the reads as paths of a dBG represented with a BF. The dBG is built from solid k-mers of the reads, that is, k-mers occurring multiple times in the reads. A read is anchored in the graph if it contains at least one solid k-mer and encoded as a list of graph bifurcations from this anchor.
1.3. Contributions
In this article, we present a new alignment-free and reference-free method, dynamic alignment-free and reference-free read compression (DARRC), that compresses the sequencing reads dynamically. The main contribution of this work is the guided dBG (gdBG) that allows a unique traversal to reconstruct the reads it was build from. The gdBG is indexed using a colored dBG succinct data structure, the Bloom Filter Trie (BFT) (Holley et al., 2016) that enables the update of the gdBG with reads of other similar genomes. Additional methods are presented to optimize the encoding of the reads. On a large Pseudomonas aeruginosa data set, DARRC outperforms all other tested tools. It provides a 30% compression ratio improvement in single-end mode compared with the best performing state-of-the-art HTS-specific compression method in our experiments.
2. Methods
A string x is a sequence of symbols drawn from a finite nonempty set, called the alphabet
2.1. The de Bruijn graph
A dBG is a directed graph
The dBG is a long-studied abstract data structure used in computational biology. It is particularly useful for the problem of read assembly (Compeau et al., 2011), in which the goal is to reconstruct a genome as a single sequence from a set of reads. For this purpose, it is necessary to find a Hamiltonian cycle in the graph, a path starting and ending on the same vertex that visits each vertex exactly once. Although heuristics exist to extract Hamiltonian cycles from a graph, the read assembly problem is yet to be solved because a Hamiltonian cycle is only one possible reconstruction of the original genome the graph was built from.
2.2. The guided dBG
The read assembly problem shows that different traversals of dBGs are possible. In the worst case, the number of possible paths between two vertices in a graph is infinite if the graph is cyclic, and exponential otherwise. Given a dBG built from a sequence and a starting vertex for the traversal, the dBG must be supplemented with information to guide its traversal to reconstruct the sequence it was built from.
A gdBG built from a sequence S is a cdBG
Encode(S, k)
Algorithm 2 decodes a sequence S from a gdBG G using vertices of length k starting with k-mer prefix x. Algorithm 1 guarantees that for any k-mer and one of its partitions, this k-mer can only have zero or one successor in the graph with the same partition. Therefore, Algorithm 2 traverses the graph by searching, for each traversed vertex, the successor with the same partition. If it is not found, the partition index is incremented and the traversal continues. As for Algorithm 1, the algorithm requires
Figure 1 represents a simple cyclic dBG built from a sequence containing a repetition. An infinite number of sequences could be extracted from the dBG because of the cycle. However, by augmenting the dBG with partitions, Algorithm 2 will traverse the cycle only once during the reconstruction of the sequence. Indeed, when Algorithm 1 tries to insert k-mer agt with partition 1, a successor with the same partition is found. Therefore, k-mer agt is inserted with partition 2 such that the cycle is not contained in one partition.
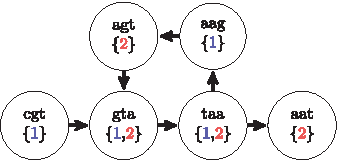
The gdBG of sequence
An important property of gdBGs using implicit edges is that no false implicit edge can be traversed during the decoding.
Proof. If a false implicit edge connects vertices not sharing a partition, Algorithm 2 will not consider this edge as only successors with the same partition are traversed. If a false implicit edge connects vertices v and
Algorithm 1 does not distinguish true implicit edges from false implicit edges, ensuring that Definition 1 is always respected during the encoding.
Furthermore, partitions allow to apply the following generalized definition of edges in dBGs to gdBGs:
For a sequence S to encode in a gdBG and

The gdBG of sequence
3. Compression
Section 2 presents methods to encode a sequence as a gdBG and to decode it. In this section, we describe how to use this methodology to compress reads. To improve compression efficiency, we preprocess the reads.
3.1. Read clustering and merging
A simple form of read assembly extended from ORCOM (Grabowski et al., 2014) is performed to reduce the input data. It clusters reads according to their minimizer, then merges reads sharing an overlap within each cluster and finally merges reads sharing an overlap but originating from different clusters. These three steps are described in the following.
3.1.1. Clustering
The minimizer (Roberts et al., 2004) of a read r is the lexicographically smallest of its p-mers with
3.1.2. Intracluster merging
Within each cluster, the reads are sorted by decreasing position of their minimizer, in which reads sharing the same minimizer position are sorted lexicographically. For each read r and its minimizer m at position pm, all reads
3.1.3. Intercluster merging
As an extension of the previous steps used by ORCOM, we additionally perform a process similar to the intracluster read described previously to merge super reads from multiple clusters. For each super read sr and its minimizer m at position pm, a new minimizer
3.1.4. Paired-end reads
Each mate of a pair is considered as a single read that is clustered and merged using the previously described methods. However, the clustering and merging steps keep track of the position of the mates in the SSRs. This information is used afterward to store in each read meta data whether the read is the first mate of its pair. In such a case, the position of its corresponding mate in the SSRs is stored as well.
3.2. SSR encoding
Encoding a set of SSRs using a gdBG requires to extract k-mers from the SSRs. If edges represent overlaps of length

Extraction of 4-mers overlapping on
To keep the growth of the gdBG small when inserting a new SSR, we determine the k-mer extraction start position, called start position in the following, which maximizes the number of k-mers already stored in the gdBG. To this end, we maintain in memory a k-mer index recording all k-mers extracted. As the cost in time and memory of such an index is prohibitive, we use a BF instead.
Introduced by Bloom (1970), a BF records the presence of elements in a set. Based on the hash table principle, look-up and insertion times are constant. The BF is composed of an array B of b bits, initialized with 0 s, in which the presence of n elements is recorded. A set of f hash functions
and
respectively, in which
Algorithm 3 (see Section 7) is a greedy approach making use of the BF to iteratively detect for each SSR of a set its optimal start position and updating the BF with all novel k-mers. The optimal start position of an SSR is a position from 1 to l maximizing the number of k-mers extracted that are already present in the BF compared with the other possible start positions. Once the optimal start position of an SSR is determined, the BF is updated with the k-mers extracted and the next SSR of the set is processed. To encode all SSRs completely, this approach does not only return the k-mers to insert into a gdBG, because these do not necessarily cover the entire SSRs. It also returns the head and tail of each SSR, which are the uncovered prefix and suffix, respectively, not encoded in the gdBG. In addition, to provide an entry point into the gdBG for the decoding, it returns the starting overlap of each SSR, which is the
3.3. Partition encoding
3.3.1. Encoding
Partition sets associated with k-mers in gdBGs are represented as lists of sorted integers. A naive way to store a partition set is to use a fixed number of bytes for each partition. For example, 4 bytes is a standard size for integers on current computer architectures. To decrease the memory footprint while keeping the lists indexed, partitions are first delta encoded by storing the difference between each integer and its predecessor in the list (or 0 if the integer is in first position). The resulting values are called deltas. However, it only decreases the minimum number of bits necessary to encode the partitions, but not their final representation. Consequently, deltas are Vbyte encoded (Williams and Zobel, 1999): each byte used to encode a delta has one bit indicating whether the byte starts a new delta or not, allowing to remove unnecessary bytes from each delta. Thus, partitions use a variable number of bytes proportional to the minimum number of bits necessary to encode their deltas.
3.3.2. Recycling
As a small delta produces a small encoding, partition integers are recycled instead of naively using the next higher integer for every new partition as, for the sake of convenience, described in Algorithm 1. Partition sets a and b can share the same partition integer if they are not neighbors in the graph, that is, no k-mer suffix or prefix of a overlaps a k-mer prefix or suffix of b, for suffixes and prefixes of length
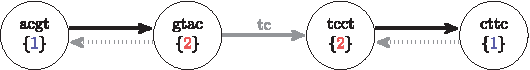
The gdBG of SSRs
As there can be a large number of partitions in the graph, verifying the connectivity of one partition to all other partitions is often impractical. We propose instead a heuristic that verifies the connectivity only to the last t partitions inserted, t being a user-defined threshold, such that these t partitions are the only candidates for recycling. Using partition recycling requires to save the partitions traversal order that cannot be incremental anymore as shown in Algorithms 1 and 2.
3.4. Meta data and gdBG compression
Steps described previously generate meta data specific to one input file such as read lengths and positions in SSRs. These meta data are first encoded in separate streams and are then compressed using an LZ-type algorithm, LZMA (Pavlov, 1999). After all k-mers and partitions are inserted in the gdBG, the latter is written to disk. As it must be loaded in memory for every update and decompression, the gdBG is compressed with Zstd (Collet, 2015), a compression method based on Huffman coding and Asymmetric Numeral Systems (Duda, 2013) that favors compression and decompression speed over compression ratio.
4. Update and Decompression
To update a compressed archive with a new input file, only the gdBG previously created is decompressed and loaded in memory, as meta data are not used for the update. A fast procedure iterates over all k-mers of the gdBG and inserts them into the BF of Algorithm 3 (see Section 7) instead of starting with an empty BF to optimize the choice of the k-mer extraction start positions in the SSRs. The gdBG is then updated with the new k-mers and partitions. The starting partition index is greater than the partition indexes already present in the gdBG, ensuring that each input file is encoded with a unique set of partitions.
Decompressing a read file starts with decompressing its meta data and the gdBG it is encoded in. The gdBG is then loaded in memory and Algorithm 2 is used to traverse the gdBG, but only following those partitions that are specific to the read file to decompress. This way, single files can be decompressed separately. As Algorithm 2 decodes SSRs, meta data are used afterward to extract the actual reads. If reads are paired-end, meta data are also used to reorganize them such that corresponding mates of the same pair are together in the decompressed file.
5. Results
DARRC is implemented in C and uses the BFT library (Holley et al., 2016) for its gdBG. The BFT provides time and space efficient functionalities that are required by DARRC. These functionalities include (i) the ability to update the BFT with new k-mers and colors without recomputing the index, (ii) k-mers extraction from the BFT, and (iii) prefix search over the set of k-mers within the BFT. The software is available at (https://github.com/GuillaumeHolley/DARRC). We compared DARRC with three state-of-the-art de novo DNA sequence compression tools: ORCOM (Grabowski et al., 2014), LEON (Benoit et al., 2015), and Mince (Patro and Kingsford, 2015). DARRC was also compared with the same LZ-type algorithm used to compress its meta data, LZMA (Pavlov, 1999). Experiments were carried out on a server with 378 GB of RAM and two 8-core Intel Xeon E5-2630 v3 2.4 GHz processors. All input files were placed on mechanical hard drives. Compressed archives and decompressed files, during compression and decompression, respectively, together with temporary files such as read clusters were written to a RAM-based partition when the tools allowed to specify an output directory.
As the current version of DARRC does not take advantage of parallelism, all software were run using a single thread, except Mince that requires a minimum of four threads. All de novo DNA sequence compression tools were run using their default parameters. LZMA was run with the same compression level as the one used to compress DARRC meta data. DARRC default parameters are minimizers of length 9 for the clustering, 5 mismatches allowed per read merging and 36-mers overlapping on 11 symbols for the gdBG. ORCOM, LEON, Mince, and LZMA compressed all files in separate archives, whereas DARRC updated the same archive iteratively with the files to compress: each iteration decompressed and reloaded the necessary data from the data written to disk in the previous iteration.
The data set used for the experiment consists of 473 clinical isolates of P. aeruginosa sampled from 34 patients (NCBI BioProject PRJEB5438), resulting in 338.61 Gbp of high coverage sequences. Reads are 100 bp paired-end reads generated by Illumina HiSeq 2000. Pair mates were placed in different files for every isolate. The experiment was run in single-end mode and paired-end mode for all tools such that in the single-end mode, every mate file is considered as a single-end read file. The appropriate single-end and paired-end modes were used for DARRC and Mince. The mates were concatenated for the paired-end experiment of ORCOM as the tool neither preserves the order of the reads nor stores the paired-end information. LEON and LZMA do not have an explicit paired-end mode but keep the original order of the reads, thus for the paired-end experiment of LEON and LZMA, the mate files of every isolate were concatenated.
Compression ratios in paired-end mode and single-end mode are shown in Figure 5. DARRC clearly outperforms all the other tested tools in both modes. In paired-end mode and single-end mode, DARRC uses about 0.261 and 0.204 bits/base, corresponding to a 57% and 30% compression ratio improvement compared with the second best results, respectively. The paired-end compression ratio of ORCOM compared with its single-end compression ratio shows that the tool is not adapted to paired-end read compression. The gdBG represents about 10% and 13% of the data written to disk by DARRC in paired-end mode and single-end mode, respectively.

Compression ratios in paired-end mode (left) and single-end mode (right).
DARRC compressed more than two times faster than LZMA but used the most time to decompress, as shown in Figures 6 and 7, respectively. DARRC's compression time overhead is explained by the fact that at each iteration, the gdBG must be decompressed, loaded in memory, and updated with new k-mers and partitions.
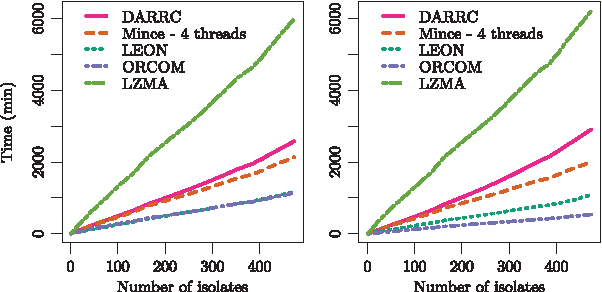
Compression times in paired-end mode (left) and single-end mode (right).
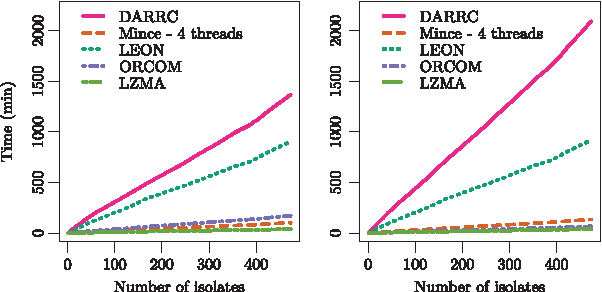
Decompression times in paired-end mode (left) and single-end mode (right).
All tools performed compression and decompression using a maximum of 4 Gb of main memory, an amount nowadays available on most desktop computers and laptops. Even by updating the same archive iteratively, DARRC compression used <2 GB of main memory.
6. Conclusions and Future Work
We presented DARRC, a dynamic alignment-free and reference-free read compression method that can incrementally update compressed archives with new genome sequences without full decompression of the archives. DARRC uses a new abstract data structure, the gdBG, that allows a unique traversal of the dBG to reconstruct the sequences it is built from. We showed that, on a large pangenome data set, our method outperforms several state-of-the-art DNA sequence compression methods and a general purpose compression tool regarding the compression ratio while achieving reasonable running time and main memory usage. Furthermore, we showed that the compression ratio of DARRC is attractive even with only few files compressed. Future work concerns the parallelization of the software, particularly the read clustering and merging phase that offers a lot of potential for multithreading. In addition, a logical evolution of DARRC is the introduction of a pattern matching functionality within the compressed data as in Yu et al. (2015), leading to large-scale complex methods such as read alignment and variant calling using multiple genomes.
7. Appendix
Footnotes
Acknowledgments
This research is funded by the International DFG Research Training Group GRK 1906/1 for G.H. and R.W., the NSERC Discovery Frontiers grant on “Cancer Genome Collaboratory,” and the NSERC Discovery Grant to F.H.
Author Disclosure Statement
The authors declare that no competing financial interests exist.