
Abstract
Abstract
Next-generation sequencing, also known as high-throughput sequencing, has increased the volume of genetic data processed by sequencers. In the bioinformatic scientific area, highly rated multiple sequence alignment tools, such as MAFFT, ProbCons, and T-Coffee (TC), use the probabilistic consistency as a prior step to the progressive alignment stage to improve the final accuracy. However, such methods are severely limited by the memory required to store the consistency information. Big data processing and persistence techniques are used to manage and store the huge amount of information that is generated. Although these techniques have significant advantages, few biological applications have adopted them. In this article, a novel approach named big data tree-based consistency objective function for alignment evaluation (BDT-Coffee) is presented. BDT-Coffee is based on the integration of consistency information through Cassandra database in TC, previously generated by the MapReduce processing paradigm, to enable large data sets to be processed with the aim of improving the performance and scalability of the original algorithm.
1. Introduction
T
The next-generation sequencing (NGS) revolution has drastically reduced the time and cost requirements for sequencing large genomes, producing massive amounts of data. So, for large-scale analysis, technical issues such as speed and scalability also become important parameters (Muller et al., 2009). High-throughput comparative analyses require automated and fast pipelines that include numerous MSAs as the starting point for structural and functional studies (Thompson and Poch, 2006) and phylogenomic approaches (Dunn et al., 2008). However, the larger and longer the sequence data sets to align are, the higher is the error introduced and the lower is the alignment accuracy. Some methods allow computation of larger data sets while sacrificing quality, and others produce high-quality alignments, but scale badly with the number of sequences and require huge amounts of time to provide the solution (Sievers et al., 2013).
Consistency-based aligners are good alternatives for minimizing the large-scale alignment errors. These methods, like T-Coffee (TC) (Notredame et al., 2000), have been demonstrated to be able to reduce the errors in the first stages of the progressive alignment, using the consistency information to take in consideration the global constraints when performing each individual pairwise alignment. These global alignment constraints are obtained through an initial stage that builds the consistency library compounded by all possible pairwise alignments among all the input sequences.
However, consistency-based MSAs have an important drawback, namely their poor scalability for use with large-scale data sets. Owing to the computational requirements of the consistency library (
Limiting the amount of consistency information triggers an important reduction in the accuracy of the final alignment. When dealing with a large-scale alignment, it is probable that no more than 5% of the consistency will fit in the local main memory. Therefore, the poor accuracy achieved in these cases makes this approach impractical for large-scale data sets.
The second alternative solution is based on increasing the storage capacity available to maintain all the consistency information even when large alignments are done. This can be done by storing the consistency on a disk, that is, in a database, and then providing more capacity. Moreover, we can use a massive data processing infrastructure to increase the storage capacity from gigabytes to petabytes.
In this article, we plan to adapt TC to the NGS, as it is the most data-intensive progressive aligner given how it generates and uses the consistency. We use the Hadoop infrastructure (Zhang et al., 2016) and MapReduce paradigm (Dean and Ghemawat, 2010) to calculate and store the consistency library. This allows an increase of the library size by more than six orders of magnitude. Our proposal is based on using the Cassandra distributed database (Carpenter and Hewitt, 2016) to store the resulting consistency library.
However, increasing the number of sequences leads to an important increase in the execution time, given that calculating the consistency library is a hugely demanding computational process. Accordingly, taking advantage of the Big Data paradigm, it is relevant to redefine the way in which the consistency library is obtained and thus enabling the final execution time to be reduced. Here we present an innovative method based on the Big Data MapReduce paradigm, applying Apache Spark to calculate the consistency library. MapReduce is a programming paradigm that allows massive computational resources (processors, memory, and disks) to be exploited in a simple and scalable way.
The proposed method has been integrated into TC, and this new version is identified as Big Data T-Coffee (BDT-Coffee). The main goal of this article is demonstrate that Big Data technologies improve the performance and scalability of consistency-based MSA tools, thus allowing reductions in the execution time and the alignment of bigger data sets.
2. State of the Art
When using the Gotoh (1982) or Myers and Miller (1988) dynamic programming techniques, it is possible to reach the optimal alignment for two sequences. Those techniques are based on scoring the residues match using a substitution matrix or a consistency library. However, for a greater number of sequences, the alignment is a nondeterministic polynomial NP-complete problem, requiring the utilization of heuristics algorithms. In this situation, the goal of the MSA is to seek an alignment that maximizes its accuracy, approximated by the sum of similarities for all pairs of sequences (Sum-of-Pairs [SP] score) (Just, 2001). Among the different MSA approaches, progressive alignment is the most prevalent heuristic method for large data sets.
Progressive alignment builds up a final MSA by combining pairwise alignments beginning with the most similar pair and progressing, following a guide tree, to the most distantly related. The most popular progressive alignment implementation is the Clustal family, Higgins and Sharp (1988), especially the ClustalW weighted variant proposed by Thompson et al. (1994), which is used by a large number of web portals. The main drawback of these methods is that errors made in early pairwise alignments are propagated to the final result, thus affecting the accuracy of the global alignment. To lessen the early error propagation, consistency-based methods were proposed.
Iterative algorithms, such as MUSCLE (Edgar, 2004), MAFFT (Katoh and Standley, 2013), and ProbCons (Do et al., 2005), overcome the greediness of progressive alignment methods through a process of alignment refinement to optimize the final result. These approaches start with an initial solution, which is improved using iterative steps. Evolutionary and genetic algorithms are an enhancement of iterative algorithms that use a stochastic process to improve the solution. Evolutionary methods start with an initial population of individual solutions and make them evolve using crossover and mutation operations to select the best individual based on its fitness (alignment accuracy). Good examples of such genetic algorithms are rubber band technique genetic algorithm (Taheri and Zomaya, 2009), multiple sequence alignment genetic algorithm (Gondro and Kinghorn, 2007), and vertical decomposition genetic algorithm (Naznin et al., 2011). Recently, a few studies have been implemented on multiobjective genetic-based methods to cope with the different fitness functions used to optimize the alignment: multiobjective optimizer for sequence alignments based on structural evaluations (Ortuno et al., 2013), multiple sequence alignment with affine gap by using multiobjective genetic algorithm (Kaya et al. (2014), and hybrid multiobjective memetic metaheuristic for multiple sequence alignment (Rubio-Largo et al., 2016).
Consistency-based methods use consistency information from different pairwise alignments to improve the final result. TC (Notredame et al., 2000) is the most representative method in this category, combining the consistency objective function for alignment evaluation (Coffee)-based scoring function (Notredame et al., 1998) with the progressive alignment algorithm. TC introduces a library generated using all-against-all pairwise alignments computed with a pair hidden Markov models to reduce greediness and increase accuracy. Other common MSA programs that use consistency are Probcons (Do et al., 2004), Probalign (Roshan and Livesay, 2006), and the LNSI variant of MAFFT (Katoh and Standley, 2013). However, such methods are severely penalized by the memory requirements needed to store the consistency information, which limits their performance and scalability.
The problem of scalability is common in many other bioinformatics tools and algorithms. Nowadays, bioinformatics is challenged by the fact that traditional analysis tools have difficulties in processing large-scale data from high-throughput sequencing (Zou et al., 2014). The utilization of high performance computing (HPC) and big data infrastructures has recently given bioinformatics researchers an opportunity to achieve scalable, efficient, and reliable computing performance on Linux clusters and cloud computing services.
The open-source Apache Hadoop project (Zhang et al., 2016) has demonstrated the ability to store and process petabytes of information in a timely and cost-effective manner. Big data technology distributes the data over commodity hardware clusters, providing scalable parallel processing and storage for performing distributed analytics of big data sets. The architecture comprises two main components: the Hadoop distributed file system (Karun and Chitharanjan, 2013) and the MapReduce framework (Dean and Ghemawat, 2010). MapReduce is a programming paradigm that expresses a large distributed computation as a sequence of fault-tolerant distributed operations on data sets of key–value pairs across the nodes of a Hadoop cluster.
Moreover, Hadoop has a complete stack of services and frameworks (Spark, Cassandra, etc.) that provide a wide range of machine learning and data analysis tools to process any workflow type. The Apache Spark is a general-purpose cluster-computing engine that is very fast and reliable (Sakr, 2017). Spark supports in-memory computing, and this enables it to query data much faster than disk-based engines such as Hadoop (100 × faster for some problems). The Apache Cassandra is an open-source distributed NoSQL database designed to handle large amounts of data across many commodity servers, providing high availability and scalability with no single point of failure (Carpenter and Hewitt, 2016). Cassandra is a hybrid between a key–value and a column-oriented data model.
Recent bioinformatics tools have already taken advantage of these new technologies to improve their performance and scalability. In the area of MSA, Sadasivam and Baktavatchalam (2010) proposed a novel approach that combines the dynamic programming algorithm with the computational parallelism of Hadoop data grids to improve accuracy and accelerate MSA. In Zou et al. (2015), the authors developed HAling, a DNA MSA tool based on trie trees to accelerate the center star MSA strategy. It was implemented using the MapReduce distributed framework. The use of the MapReduce paradigm and Hadoop infrastructures enabled the scalability and the alignment time to be improved. In Wan and Zou (2017), a new version of this tool was implemented using the Apache Spark framework. The Smith–Waterman algorithm for protein sequence alignment and the neighbor-joining method for phylogenetic trees construction were implemented in Spark, showing a high memory efficiency and good scalability. PASTASpark (Abuín et al., 2017) uses Apache Spark to boost the performance of the alignment phase of PASTA, which is the most expensive task in terms of time consumption.
Based also on Spark framework, Wiewiórka et al. (2014) developed SparkSeq, a general-purpose tool for RNA and DNA sequencing analyses, tuned for processing in the cloud big alignment data with nucleotide precision. Zhao et al. (2015) developed SparkSW, which can carry out the Smith–Waterman algorithm in load-balancing way on a distributed system to cope with increasing sizes of biological sequence databases. In SparkScore (Bahmani et al., 2016), a set of distributed computational algorithms is implemented in Apache Spark, to leverage the embarrassingly parallel nature of genomic resampling inference on the basis of the efficient score statistics.
There are more MapReduce solutions in the area of search and mapping short reads against a reference genome. These applications, CloudBlast (Matsunaga et al., 2008), CloudBurst (Schatz, 2009), SEAL (Pireddu et al., 2011), and CloudAligner (Nguyen et al., 2011), implement traditional algorithms such as Blast (McGinnis and Madden, 2004), RMAP (Smith et al., 2009), and BWA (Li and Durbin, 2009) using the MapReduce paradigm.
3. Method
In Lladós et al. (2017), we presented probabilistic pairwise model for consistency-based multiple alignment in Apache Spark (PPCAS), an innovative method able to generate the parallel probabilistic pairwise model for large data sets of proteins and also store this in a distributed platform using the TC format. To use the external library generated, it was passed by file into the TC tool. However, the problem of aligning thousands of sequences was there, as the library loaded into the main memory to proceed with the dynamic programming, those huge amounts of consistency could not fit onto it.
To solve this problem, we had to change the paradigm. Thus, we used Apache Spark to calculate the consistency and then store it into the NoSQL Cassandra distributed database. The next step was to integrate it into TC using the Datastax connector, which allows access to the database through the network. This new structure, named BDT-Coffee, can run on a modest desktop computer, while it can access the enormous resources of the Cassandra nodes to speed up the alignment.
This approach has several advantages. First, it solves the consistency storage problem. The Cassandra database uses the hard disk as the main consistency library container, so the capacity is much larger than the main memory, and then allows TC to perform the alignment regardless of the number of sequences.
The next advantage comes from the fact that Spark is a really fast engine for large-scale data processing in real-time executed over Hadoop. Spark has a master/slave architecture. It has one central coordinator (driver) that communicates with many distributed workers (executors). In this structure, the driver is the process where the main method runs and the executors are those that process the data received. It is also important to note the efficiency, adding more nodes to the infrastructure will ensure that the execution time of the program will decrease.
However, the critical point is to deal with the latencies due to the consistency information access, which is stored in Cassandra. The original version of TC uses the primary library, which is placed in memory, for building the alignment. The dynamic programming (Myers and Miller, 1988) performs a large number of accesses to the library to align while it follows the guide tree. The number of accesses is quite high and it grows enormously based on the number of sequences and their length. It is obvious that moving all these accesses to a disk connected through the network is going to be slower than accessing the main memory.
Apache Cassandra is an open-source distributed database system that is designed to store and manage large amounts of data across commodity servers. Cassandra can serve as both a real-time operational data store for online transactional applications and a read-intensive database for large-scale business intelligence systems.
Therefore, TC executes a large number of accesses to build the alignment. It implies that our method has to do the same as them for the same data set. This means that we have to spend more time by reading from the Cassandra database plus the latency.
To tackle the final development, we identified the problem in three different subtasks: (1) the library calculation, (2) the storage of the library to be easily accessed, and finally (3) the integration of the library with TC.
3.1. Library calculation
It is important to note that Spark only supports Java, Python, or Scala programming languages. Owing to this fact, we have to implement the library calculation in Python, which directly affects the performance. This implies that the Python/Spark library computation will perform worse than the TC version, which is written in C.
Owing to this problem, we decided to wrap the probabilistic pairwise model code into a compilable and executable Python code by using Ctypes. Ctypes is a native included library (since Python 2.5) that allows us to use shared functions and libraries from C (and whichever OS) and to obtain the performance of C language without renouncing the simplicity and portability of Python.
The MapReduce paradigm is used to parallelize the consistency calculation tasks. This allows BDT-Coffee to compute the primary library through as many nodes as the Hadoop cluster has. In the implementation, the map stage is responsible for defining all the tasks in charge of computing the probability score for a set of pairs of sequences.
In Algorithm 1, the driver generates these tasks for all the
Spark parallel pairwise probability calculation
The executor, lines 4–9, performs a subset of the pairwise combinations. This is done in the double-nested loop in lines 4 and 5, which obtains the different combinations of sequences assigned to the task. It calculates the library for each of these combinations by calling the pair_wise(Si, Sj) function of the shared library (PPCAS.so). This function calculates the probabilistic pairwise model for these two sequences and sends it back to the driver.
3.2. Library storage
After generating the library in Apache Spark, we need an interface to read and write the data efficiently. The library is composed of tuples (Si, x, Sj, y, and
Apache Cassandra is the database chosen as the interface in our application. It is a key–value NoSQL database with peer-to-peer distributed architecture and massively scalable. Therefore, by using the Cassandra database, we are able to reduce the latency as it is lower than a plain file disk latency.
A connector between Spark and Cassandra database is used to write the primary library into Cassandra. This connector is the datastax:spark-cassandra-connector, which allows a whole DataFrame to be inserted into Cassandra efficiently.
Figure 1 shows the schema used in Cassandra. As with all the databases, Cassandra does not allow repeated keys in their key spaces. This is a problem, because many keys “Si x” and “Sj y” are repeated across different sequences and residues in the library. To solve this, we use a partition key (PK) for the key “Si x” and a clustering key for key “Sj y,” so we could have multiple repetitions of the PK. The PK is also responsible of the locality of the data, so all the constraints with the same PK will be in the same node.
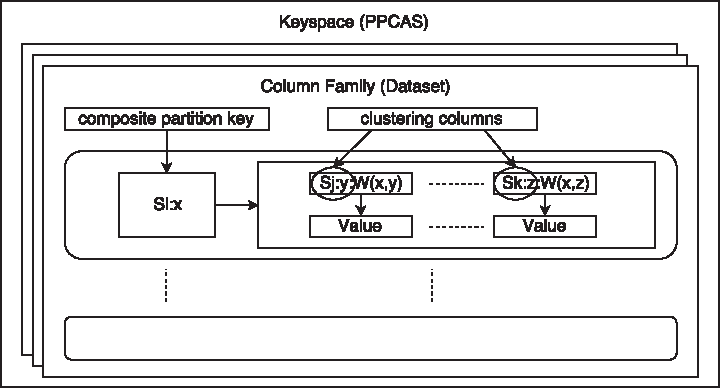
Database schema used by PPCAS.
3.3. Integration with TC
A connector between C code and Cassandra database is used to communicate Cassandra with TC. This connector is the Datastax C/C++ connector that allows the primary library generated by PPCAS to be requested.
The execution times of the first test generated by the integration of TC and PPCAS with Cassandra database were extremely slow. The number of queries to Cassandra was extremely high, a small data set of four sequences with an average length of 90 residues, their execution time multiplied by 100, from 0.02 to 2 seconds. Thus, the growth increases exponentially when more sequences are aligned. We have to note that there were many repeated queries among the alignment, because when two sequences are aligned a profile is generated, and this profile is aligned with another sequence/profile, requesting all the data used to align the first two sequences, and this propagates throughout the progressive alignment.
To avoid repeated queries to Cassandra, we decided to implement two levels of dynamic cache, the consistency cache (CC) and the score cache (SC); see Algorithm 2. The first stores the data received by each query in the memory and the second stores the scores of the extension process. Thus, the second and subsequent times that a primary library data related to a sequence–residue are needed by the program, it checks whether the extended score is available on the SC. If not, it checks in the CC to build the extension before querying Cassandra. Using this technique also implies a limited cache memory. Hence, if the whole library is bigger than the free main memory of the node running TC, a replacement policy is needed.

Therefore, the replacement policy has to be aware of the free memory available in the node, and it pushes out as many constraints held by “Si x” as needed to fill the size of the new one. Different approaches were tried to selecting which sequence–residue to delete. First, a random strategy, which provided a high miss rate. Next, we implemented a least recently used policy. This improved the hit ratio of the cache at the expense of higher memory usage. Finally, the first in, first out (FIFO) strategy was evaluated, providing a better balance between the hit ratio and the memory requirements. This strategy uses a queue in which a constraint is pushed each time a sequence–residue is used, and it pops out the oldest one when a replacement is needed.
This implementation reduced the huge latency generated by so many queries to Cassandra. To optimize the latency even further, we decided to group the data by its residue. So, the data stored in the library by a key “Si x” will become “Si x/chunk”, the chunk being the number of keys indexed together. An example of this optimization is shown in Figure 2. When a query asks for “Si x,” it would not only retrieve all the constraints of this PK but also ask for all the “Si x/chunk” data. In the example, we reduce the number of required accesses to Cassandra by 5 (chunk size). Furthermore, to rebuild the original “x” value, we add it as a new clustering key in the column family.
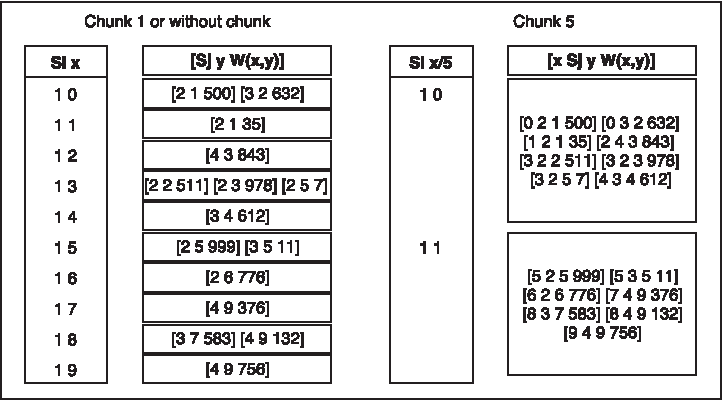
Use of the chunk optimization.
Using this, we can reduce the number of queries by chunk. This is because when TC asks for a key, whether or not it is in the CC, it has to make a query to Cassandra. Adding the chunk, we can retrieve and store multiple data related to a sequence–residue pair range into the CC. When this extra data are needed, it is already in the CC.
4. Experimentation
In this section we evaluate Big Data Tree-based Consistency Objective Function For alignment Evaluation
Both benchmarks are briefly introduced:
BALiBASE (Thompson et al., 2005) is a database of high quality documented and manually refined reference alignments based on 3D structural superpositions. The accuracy of the alignments is measured using two metrics: the SP and the total column score, which are obtained by comparing the user alignment against a reference alignment. HomFam (Sievers et al., 2011): The existing benchmark data sets are very small (150 and 50 sequences in BALiBASE and Prefab, respectively). Homfam provides large data sets using Pfam families with thousands of sequences. To validate the results of aligning a Pfam family, the Homstrad site contains some reference alignments and the corresponding Pfam family. These references are previously dealigned and shuffled into the data set. After the alignment process, the reference sequences are extracted and compared with the originals in Homstrad.
HomFam contains almost 100 sets. We selected the protein family named rrm to evaluate the method. The results for the execution time presented in this section represent the average results obtained after evaluating the corresponding family. Furthermore, each experiment corresponds to five iterations to show the robustness of the results.
The execution environment was an Apache Spark cluster made up of five nodes, each one characterized in Table 1 for a total of 80 cores and 320 GB of memory. All the test running PPCAS made use of 80 execution cores and the default memory (usually 512 MB for each core). Meanwhile T-Coffee and BDT-Coffee were limited to one execution core and 8 GB of RAM.
4.1. Chunk: reducing the number of accesses
To achieve faster execution times, it is important to reduce the number of queries to the database. In this section, we use different chunk configurations to validate them and obtain an optimal chunk for the incoming tests.
Figure 3 shows each chunk sizes (1, 10, 100, and 200) for each number of sequences (100, 200, and 500). As the chunk number increases, the execution time is reduced, until the maximum groupings of chunk are reached. This is due to the residue length of the aligned family. Another data set with more residues may reach its maximum groupings later. In this case, going from chunk 1 to chunk 10 with rrm_500 implies a 10-time reduction in communications and 110.5 seconds less of execution time, whereas reducing to chunk 100, it reduces the time by 35 seconds more. Increasing it further would not benefit the aligner. We can observe that when the number of sequences is rather small (100–200), all the chunks achieve a similar execution time, but the chunk-100 is the fastest. In our tests, we also noticed that using big groupings (the bigger the chunk, the bigger the grouping), Cassandra can lead to time-out requests. So we recommend using chunk 10 for small data sets and chunk 100 for larger data sets.
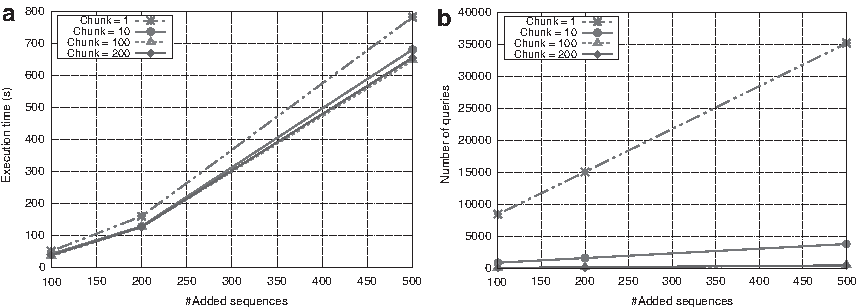
Chunk size performance with rrm family.
4.2. Consistency cache versus score cache
The cache of BDT-Coffee is limited by the main memory of the computer. In this section, we show the behavior of the cache and look for an optimal balance between CC and SC. For this purpose, the rrm family with 100 sequences is used and the chunk is set at 10.
The original execution time of TC used to align the rrm with 100 sequences is 41.202 seconds. This time should be our reference, as we do the same computations plus the queries to Cassandra. Figure 4a shows the execution time to align the set using only CC and varying the memory assigned for caching. When the cache is large enough to fit all the constraints (40 Mb), the alignment takes 43.563 seconds (2.361 in communications). As we supposed, when the size of the CC is smaller than the amount of consistency, the application became slower. In those cases, the method has to use the replacement policy and this implies more queries, as the progressive alignment uses all of them in the last step.
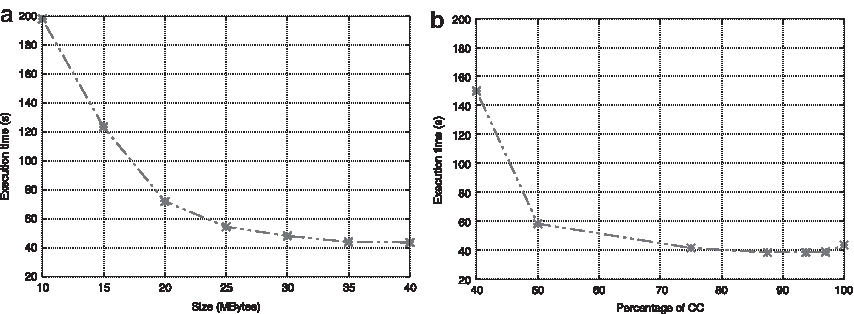
Performance of CC and SC cache memory distribution with rrm_100 data set.
Next, we add the SC to the equation. The trouble lies in finding a good balance between both caches, as they have to deal for the same memory resources. Figure 4b shows the different percentages for each one. It is important to note that the CC needs more allocation than the SC. Starting with the CC:75-SC:25, the combination beats the previous execution time using only the CC, and improves it even further beating the original execution time. Given that the best times appear between CC:87.5-SC:12.5 (38.4 seconds) and CC:93.5-SC:6.5 (38.37 seconds), we set the optimal values at 90% of the memory for the CC and 10% for the SC.
Finally, in Table 2, we evaluate the optimal setup with the one using only the CC while reducing the size of the caches. It is clear that using both caches is always better for the computation time.
CC, Consistency Cache; SC, Score Cache.
4.3. Big data tree-based consistency objective function for alignment evaluation solving BALiBASE benchmark
After finding the optimal values for the parameters in BDT-Coffee, an initial validation of the method was needed. For this reason, BALiBASE, a well-known benchmark in the field was used. Given the size of the data sets, we set the chunk size at 10. The figures are the total SP produced using the bali score. The first column indicates the MSA tool used. The results for BAliBASE subgroupings are in columns 2 to 7. Finally, the last column refers to the average score over all the families.
The results in Table 3 demonstrate that BDT-Coffee is able to obtain an equivalent accuracy. The slight differences in accuracy are due to the fact that, unlike BDT-Coffee, TC removes the smallest weighted library. In terms of execution time, BDT-Coffee is faster than TC as it uses PPCAS to generate the library and also the SC seems to improve somewhat the progressive alignment step.
TC, T-Coffee; SP, sum-of-pairs.
4.4. Scalability study increasing the number of sequences
In this section, we evaluate the rrm family with BDT-Coffee and compare it with TC. The chunk used is set to 100 as longer sequences imply more queries. Figure 5 shows the accuracy and average execution time for each test. Although BDT-Coffee is able to align up to 5000 sequences, TC crashes at 1000 as it fills the main memory (8 GB). Regarding the accuracy, shown in Figure 5a, we noted that both methods behave almost exactly, because the scoring methodology is the same.

Scalability of BDT-Coffee with rrm data sets.
The most important to highlight are the execution times. In Figure 5b, we can observe the duration of generating the library and aligning it with each method separately. As can be seen, the library generated by BDT-Coffee is much faster than in TC. This is due to our method using the Spark infrastructure. When the number of sequences is low (100–200), the speedup is not good enough, although the lack of parallel work mitigates the infrastructure performance. However, with a large number of sequences (500–1000), it achieves good values, 9.12 × and 17.3 × , respectively.
Before implementing BDT-Coffee, we thought that the method would have a very severe impact at the aligning stage. The added communications produce an implicit overhead in the execution time. Contrary to this, the alignment time also improved over the original method. Thanks to the caches and the chunk optimizations, the overhead is mitigated. Finally, we can say that the method presented is faster and can scale in comparison with the original method.
5. Conclusions
In this article, the authors present a scalable MSA tool based on TC to compute a final alignment. This method, named BDT-Coffee, uses PPCAS to calculate and store the consistency of a data set in a disk, improving the execution time and scalability of the original method. The consistency is calculated in parallel using a Spark distributed infrastructure and stored in a Cassandra database. The consistency constraints can be accessed quickly and efficiently, thanks to the proposed chunk groupings and the caches, mitigating the accesses to a slower support.
The experimentation proved that the alignment accuracy remains the same as the original method, while the execution time of TC is considerably reduced. The library calculation and writing in Cassandra achieves most of the benefit, as the MapReduce implementation provides a linear speedup as more nodes are added, being 17.3 × with 20 nodes aligning 1000 sequences. The aligning stage is where the queries to the database are made, but surprisingly with the use of the chunk and the caches, they are also able to beat the aligning time of TC with an almost 3.5 × speedup with 1000 sequences. Both speedups have the tendency to increase as more sequences are aligned, so the results are more than satisfactory. Regarding the good time results, we also need to note that the number of sequences that our method can process is greater than in the original method with the same memory resources, being able to align large data sets.
In the future, regarding BDT-Coffee, we aim to generate all the extension process on Apache Spark in advance, restricting all the computing work to the Hadoop infrastructure. This would speed up the alignment stage greatly. In contrast, we are open to implement solutions similar to other consistency tools such as in MAFFT or ProbCons, which is not trivial, as the implementations and use of the consistency are quite different.
Footnotes
Acknowledgment
This work was supported by MEyC-Spain under contract Nos. TIN2014-53234-C2-2-R and TIN2017-84553-C2-2-R.
Author Disclosure Statement
The authors declare that no competing financial interests exist.