
Abstract
Abstract
1. Introduction
G
Eukaryotic transcripts are more complex than prokaryotic transcripts as the gene sequences of eukaryotes contain noncoding regions called introns and coding regions called exons. The promoter regions of mammals and Drosophila melanogaster have been widely studied, whereas the core promoter structures of some important unicellular eukaryotes, such as budding yeast (Saccharomyces cerevisiae) and fission yeast (Schizosaccharomyces pombe), have not been comprehensively analyzed to date (Li et al., 2015). S. pombe is a unicellular eukaryote and has been subject to extensive experimental research in the study of basic principles of cell cycle regulation and cell division that can be used to understand more complex organisms like mammals and in particular humans. It contains one of the smallest number of genes yet recorded for a eukaryote, namely, 5118 (Wood et al., 2002, 2011).
In the past 20 years, numerous de novo motif discovery tools have been developed. These tools predict motifs using only a computational model without extrinsic comparison to existing data. Motifs can be found computationally and then confirmed by experiment, making the process of motif identification significantly more efficient. For example, Multiple EM for Motif Elicitation (MEME) (Bailey et al., 2009) is the most commonly used tool in this area and uses multiple expectation maximizations to discover possible motifs within the input data. Weeder (Pavesi et al., 2004) exhaustively enumerates possible consensus sequences determined from precomputed frequency files and evaluates each one. However, a survey of DNA motif-finding algorithms (Das and Dai, 2007) revealed that most tools have been shown to have better performance in lower organisms such as yeast but worse in higher organisms. For the previous motif-finding tools, early studies (Tompa et al., 2005) indicated that the highest sensitivity and precision they can achieve are 13% and 35%, respectively. One reason for the low performance is that, when trying to find overrepresented patterns from a set of sequences, patterns always contain mutations, insertions, or deletions of nucleotides.
So, motif finding remains a challenging area. Besides the problem of low accuracy, another problem is that many older motif-finding tools for promoter analysis no longer exist as software that is maintained and can be downloaded. Before next-generation sequencing (NGS) was introduced, algorithms for motif finding were primarily designed for promoter analysis, which involved a small set of sequences with hundreds of base pairs (bps), whereas motif finding for ChIP-Seq sequences usually work on large genome-wide DNA data sets with short sequences. Even though we are now in the NGS era, promoter analysis is still needed. However, as mentioned, we find that many motif discovery tools for promoter analysis are no longer available. Specifically, most currently available motif-finding tools are designed for ChIP-Seq sequences, such as DREME (Bailey, 2011) and RSAT peak-motifs (Thomas-Chollier et al., 2011). Finally, most ensemble pipelines are designed to take in FASTA for ChIP-Seq data only. We concluded that an ensemble tool with greater sensitivity for a small number of longer sequences is desirable.
Since individual tools can be better suited for specific scenarios, ensemble tools that combine the results of many algorithms should be able to yield a more confident and complete result. According to the results of the study of Wijaya et al. (2008), which tested against the data sets of Tompa et al. (2005), ensemble motif-finding tools show better performance compared with individual tools. The idea behind ensemble motif discovery is to run multiple existing tools and to combine their results to generate the final output. The idea of the ensemble approach has been widely applied in several biological areas, such as gene prediction (Aggarwal et al., 2003) and protein domain prediction (Saini and Fischer, 2005). In the past 10 years, several ensemble tools for motif finding have been developed, combining different algorithmic methods, such as MotifVoter (Yang et al., 2010), GimmeMotifs (van Heeringen and Veenstra, 2010), and EMD (Hu et al., 2006). Some of these tools rely on a hidden Markov model (Ellrott et al., 2002), such as GimmeMotifs and MEME-ChIP (Machanick and Bailey, 2011). Additionally, some compare the results against a database, such as JASPAR [Melina II (Okumura et al., 2007)] or STAMP (Mahony and Benos, 2007). It has been demonstrated that the ensemble strategy for the motif discovery problem can effectively improve both sensitivity and accuracy of the prediction (Das and Dai, 2007).
After applying multiple algorithms, it is a challenge to combine the results from those different algorithms, which are based on different scoring and evaluation systems. For example, Melina II (Okumura et al., 2007) compares the results against the JASPAR database. It does not perform any statistical analysis on the motif results, and ensembles like SCOPE (Carlson et al., 2007) rank the top motifs from component tools based on a scoring function. EMD and MotifVoter (Wijaya et al., 2008) proposed a clustering-based ensemble considering the binding sites when combining results and ranking top motifs from several algorithms.
In this study, we develop the Motif Combining and Association Tool (MCAT), an ensemble pipeline to find a single or multiple short motifs in the upstream (or promoter) regions of target genes believed to have common biological function. The development of MCAT was initially motivated by the study of cell division in fission yeast. Cell division is a highly orchestrated process. Thousands of proteins are involved, with hundreds being important regulators. People want to understand how such a complex event can be reliably executed (He et al., 1997; Kim et al., 1998). Specifically, by using S. pombe (fission yeast) as a model organism, we want to discover those motifs that are also hopefully TF-binding sites existing in the promoter region of S. pombe, which regulate the mitotic checkpoint signaling procedure (Heinrich et al., 2013, 2014).
The motifs do not necessarily appear exactly once in every sequence; one motif might appear one or multiple times in some sequences and not appear at all in others. MCAT searches for motifs by running six well-established motif discovery tools [MEME (Bailey et al., 2009), BioProspector (Liu et al., 2000), Contrast Motif Finder (CMF) (Mason et al., 2010), DECOnvolved Discriminative motif discovery (DECOD) (Huggins et al., 2011), Weeder (Pavesi et al., 2004), and XXmotif (Luehr et al., 2012)] on promoter sequences independently, then combines and reports the results using a novel algorithm we developed called VoteRank. We demonstrate that the MCAT approach provides an effective algorithm for improving sensitivity and accuracy of the prediction; see Section 6. The MCAT software is available at https://gith.com/yanshen43/MCAT
2. Problem Definition
The motif-finding problem is defined below. In detail, we consider DNA sequences over the fixed alphabet

An example of PWM. To construct a PWM, the first step is to build PFM by adding up the elements at each position. The next step is to convert PFM to PWM using log likelihoods. The highlighted scores are the highest scores at all positions. The sequence score is calculated by summing the highlighted scores. When given a threshold, the short sequence is identified as match if the sequences score is higher than the threshold. PFM, position frequency matrix; PWM, position weight matrix.
where
A particular site is evaluated by summing the elements from the scoring matrix at each position and comparing the sum to a match threshold. Sequence logo (Crooks et al., 2004) can be applied to visualize PWMs.
3. The MCAT Algorithm
MCAT is based on the generation of a pool of short nucleotide sequences coupled with position votes and refinement. The MCAT pipeline consists of four stages: a masking stage, a tool extraction stage, a combining stage, and a scoring stage (Fig. 2). MCAT executes each component motif-finding tool independently and combines their results based on the VoteRank results with comparison scores, z-score, and p-value. MCAT also provides visualization of these results (see Section 4.4).

Architectural outline of the MCAT. MME, BioProspector, Weeder, DECOD, CMF, and XXmotif are the motif-finding algorithms. Masked DNA sequences were sent to each algorithm and MotifRank will vote and refine motifs. The outputs of each motif will be shown with corresponding site locations. MCAT, Motif Combining and Association Tool.
DNA sequences may contain regions with highly biased distributions of nucleotides, which are called low-complexity regions. Low-complexity regions, such as simple tandem repeats and AT-rich regions, can lead to high-score results that are biologically uninteresting. During the masking stage, the input sequences are filtered for low-complexity regions by using DUST (Morgulis et al., 2006). DUST assigns a complexity score for each nucleotide. Any filtered nucleotides are then replaced with the character N to indicate that they should not be considered as part of a motif.
In the tool extraction stage, once the low-complexity regions are found and masked, the masked sequences are sent to each component motif-finding tool, which run independently. Each tool reports a set of candidate motifs and their respective sets of binding sites. Among all the candidate motifs, only some of them are strong motifs, and not all tools report motifs in precisely the same locations. MCAT wants to retrieve and cluster the motifs that are given by as many motif-finding tools as possible based on their votes. For the combining stage, MCAT combines independent results from each motif-finding tool with our novel VoteRank algorithm. VoteRank is described next.
VoteRank is the key new algorithm with the MCAT system. It accepts candidate motifs as votes from each motif-finding tool, ranks them, and combines motifs. It is a position-based algorithm that uses candidate motifs generated by any number of discovery tools and combines the identified regions to generate a smaller number of consensus motifs that fairly represent the significant results of all the tools.
Suppose the goal is to find the top K motifs. K is usually set to be 5 in our experiments. An overview of VoteRank is presented in Figure 3. First, VoteRank conducts a poll, letting each tool vote iteratively for the positions where it found a motif, so as to count the score of results found at each position in each sequence. As each tool usually reports results of a different number of motifs, instead of giving all candidate motifs equal weight, VoteRank can bias selection based on tool performance and the number of outputs.
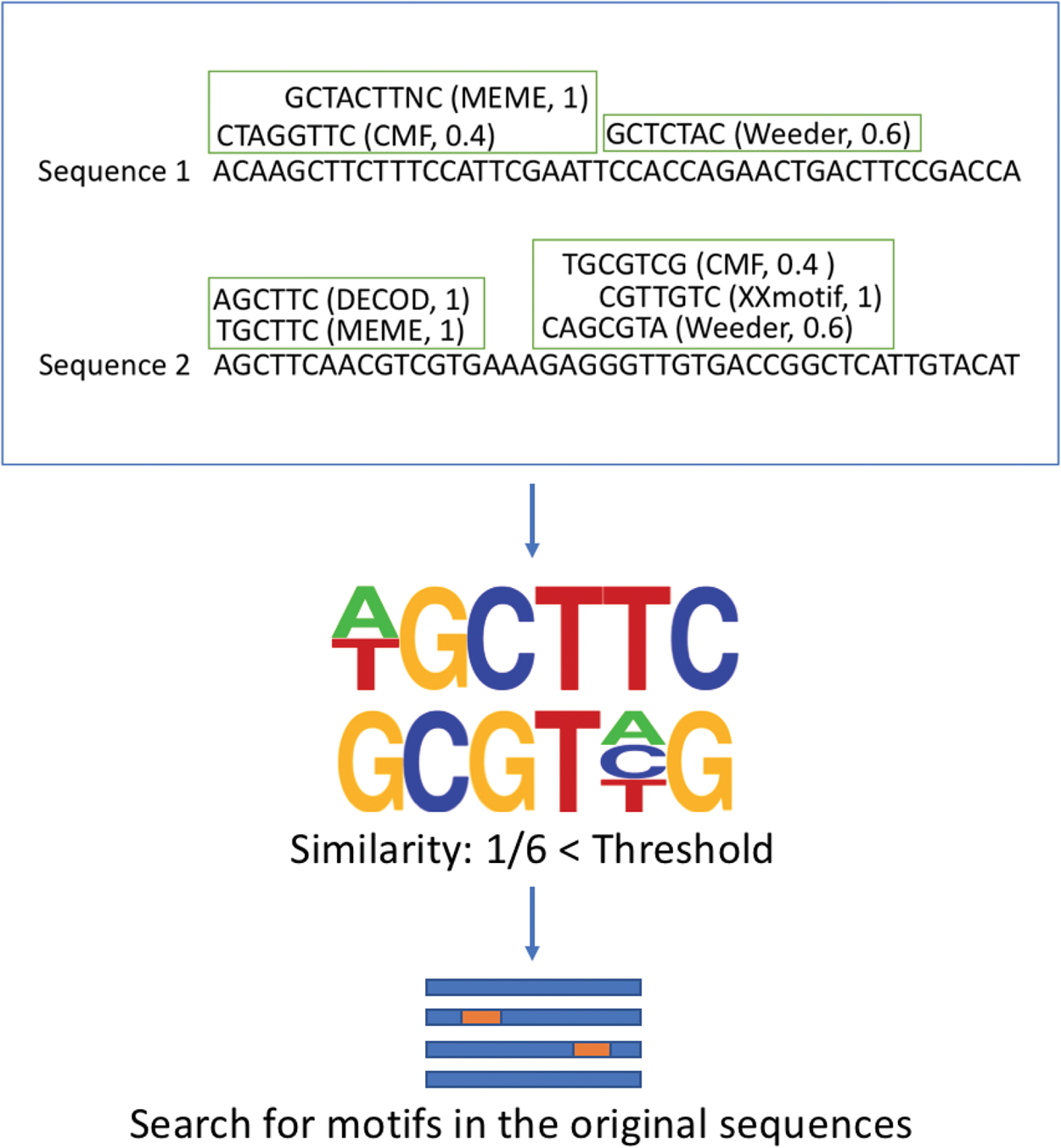
The figure shows the procedure of VoteRank. Each candidate motif votes iteratively for its corresponding position. The numbers following each motif represent their weights. For the heavily voted motifs, shown by the framed motifs, PWMs will be generated and a threshold is set to refine the poll. If the similarity is smaller than the threshold, VoteRank will verify the motifs by going back to the original sequences and searching for the motifs.
VoteRank goes through the previously conducted poll to compile the best PWMs of the motifs with the most votes and refines the motifs based on the PWM scores. For a particular sequence, suppose that two motifs x and y within that sequence come from two different tools,
The last step in VoteRank is to take the K top results and search for them in the original FASTA sequence files. It does this by looking at every position in each DNA sequence. Comparison scores are generated during this process by comparing each motif (and associated list of positions) with each motif found by a different tool and summing the results of each comparison. VoteRank will return any instances it finds that have one of the top K comparison scores to the motif. For motifs that are well conserved, this can return a surprisingly large number of results.
In the fourth and last stage, MCAT determines whether the motif is meaningful. To determine whether a motif is meaningful, one straight-forward approach is to calculate significance scores. The scoring stage represents the process of calculating comparison score, z-score, and p-value (see Section 4.3). As mentioned above, the comparison score of each motif is calculated against each other motif. VoteRank ranks all the motifs by comparison scores. Then, to further support the results, VoteRank computes statistical scores, including z-score and p-value for each of the top motifs.
4. Methods
4.1. Performance measurement
For performance evaluation, we use nucleotide-level accuracy to measure the prediction accuracy, as defined in a previous study (Hu et al., 2005).
According to the scoring schema (Zare-Mirakabad et al., 2009), index n represents that the assessment is at the nucleotide level. nTP is the number of true nucleotide-binding site positions shown predicted as binding site; nFN is the number of target nucleotide-binding site positions in known sites but not in predicted sites; nFP is the number of nucleotide-binding site positions not in target-binding sites but in predicted sites; nTN is the number of nucleotide-binding site positions in neither target-binding sites nor predicted sites.
We can define sensitivity as
We also have the nucleotide level correlation coefficient (nCC) as defined below (Burset and Guigo, 1996):
Inspired by the motif level success rate score (Hu et al., 2005), we introduced the motif level prediction accuracy (mPa) to evaluate performance of MCAT. mPa is defined as the number of target motifs Nt, which match the predicted binding sites, divided by the total number of target motifs Mt.
A prediction is considered to be a match when the predicted motif overlaps with the target motif by at least 75% of the length of the target motif.
4.2. Motif finding-tools in MCAT
The MCAT ensemble algorithm combines six tools. The details of the six tools are introduced below. These algorithms are based on different ideas, which make combining results more reliable. Note that CMF, Weeder, and XXmotif do not allow the user to specify the desired length of a motif or the number of outputs.
MEME (Bailey et al., 2009) is one of the most widely used tools for motif finding. It uses multiple expectation maximization (EM) algorithms to elicit motifs from the input data. It records each result, then masks it out, allowing MEME to find additional motifs without being distracted by motifs that have already been found.
BioProspector (Liu et al., 2000) is also a popular tool. It employs Gibbs sampling to find motifs. This is a randomized algorithm as opposed to the deterministic EM algorithm used in MEME, and the implementation in BioProspector allows for gapped motifs and motifs containing palindromic patterns.
CMF (Mason et al., 2010) is a content-based method that is designed to discriminate between two sets of DNA sequences. It first utilizes motif seeding to contrast a set of sequences believed to contain the desired motif along with a negative set and then iteratively updates seeds and generates PWMs.
DECOD (Huggins et al., 2011) uses a discriminative approach to solve the problem of motif finding. Through deconvolution and heuristic hill climbing, DECOD attempts to search for a PWM that maximizes the target function. The discriminative approach provides a significant difference in the methodology of this tool in comparison to the others, which allows the pipeline to be even more comprehensive.
Weeder (Pavesi et al., 2004) exhaustively enumerates possible consensus sequences determined from precomputed frequency files for the species, evaluating each one against the sequences from the FASTA file. It does this multiple times with differing motif lengths and number of substitutions allowed, then reports the top results.
XXmotif (Luehr et al., 2012) is an exhaustive, weight matrix-based motif discovery Web server that has a masking stage, a pattern stage, and a PWMs stage. First, it masks repeat regions and regions of local homology. Then it calculates enrichment p-values for 5-mer and 6-mer degenerate seed patterns, extends the best one, and merges similar PWMs until the p-value cannot be improved anymore.
4.3. Motif scoring
A comparison score, a z-score, and a p-value of a matching motif to a set of candidate DNA sequences are computed for each of the candidate motifs. These scores indicate how strongly the various tools were in agreement about this result. MCAT also reports the log likelihood for the PWM of each motif based on the background nucleotide ratios in the FASTA file (Stormo et al., 1982).
4.3.1. Comparison score
Given the list of found motifs, each associated with the tool it was found by, as well as a list of positions, a comparison score is created as follows: Compare each motif (and associated list of positions) with each motif found by a different tool and sum the results of each comparison. These individual comparisons are performed by finding a weighted edit distance between the two motifs, adding a constant multiplied by the reciprocal of the minimum distance between instance positions, and dividing by the square root of the length of the shorter of the sequences. A comparison score by itself only reflects on the motif quality compared with other comparison scores from within the same run of the pipeline but, taken together, can indicate how strongly the various tools were in agreement about this result. This method is mainly based on the similarity of motifs but also accounts for motifs that are reported to appear in similar locations and does not falsely weight longer motifs.
4.3.2. A z-score
In general, to compute the z-score for a sequence, one collects sequence alignment scores of one sequence to another random sequence. Then one computes the average score and standard deviation. In MCAT, motifs are sorted by the z-score. The z-score is defined as
Here, score refers to the alignment score of these sequences to other random sequences. The z-score reveals the significance of predictions based on their overrepresentation. Higher z-scores are better, because the further the real score is from the mean, the more significant it is.
4.3.3. A p-value
To support the result of VoteRank, we evaluate each output motif against all the input sequences by p-value with QFAST (Bailey and Gribskov, 1998). A p-value is defined as the probability that an event occurs by chance. The range of p-value is between 0 and 1, and the smaller the p-value is, the more significant the motif is. The original article combines p-values from independent sources in the motif-finding problem. They computed the product of position p-values and sequence p-value, then computed the confidence of this product using the QFAST algorithm as the combined p-value.
Here, MCAT takes one query motif and compares it to each sequence of the target sequences. MCAT first finds the position in each of the target sequence that best matches the motif and calculates the position p-value, which is the probability of observing a match score at least as good from a random sequence. Then the position p-value is normalized for the length of the sequence as the sequence p-value. All the independent p-values are multiplied together as the product and the combined p-value is the probability that a motif has a score better than the current product based on the distribution of the product.
4.4. Visualization
WebLogo (Crooks et al., 2004) provides the functionality in MCAT for creating sequence logos from the resulting position weight matrices generated by the pipeline. Additionally, a simplified motif location diagram is included along with the sequence logo in a generated HTML document, which displays the sequence name, the specific motif match, and the location of the motif match within the original sequence. One example of visualization is shown in Figure 4.
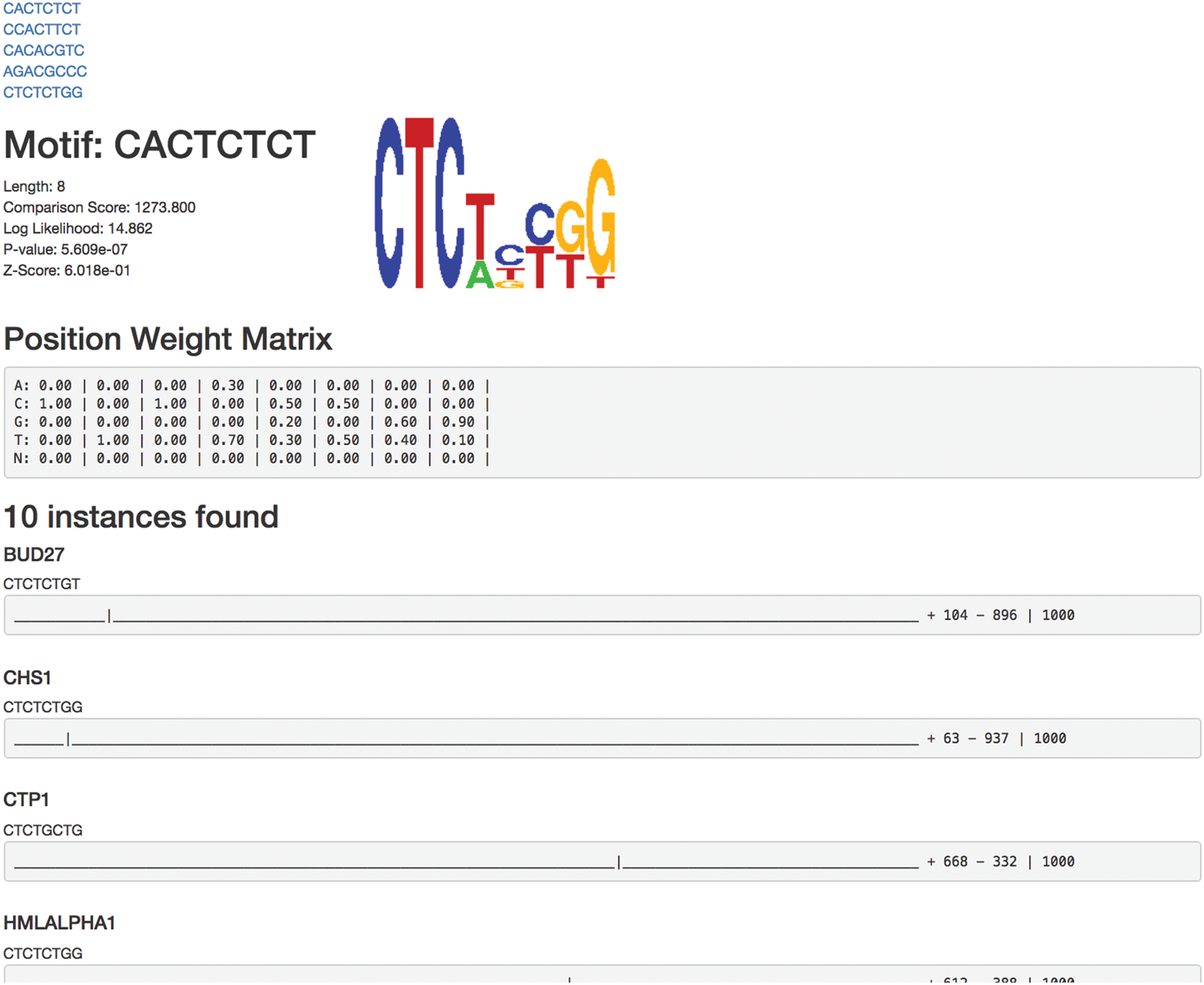
Visualization of one result. The HTML file first shows the top n motifs reported by MCAT. Then, it provides the information about each motif, including motif sequence, WebLogo, statistic scores, PWM, and positions at input sequences.
5. Comparison with Existing Tools
We compared MCAT with two other ensemble tools, EMD (Hu et al., 2006) and DynaMIT (Dassi and Quattrone, 2015), by running data sets, including S. cerevisiae data sets, S. pombe data sets, and the data sets from Tompa et al. (2005).
One ensemble tool, EMD, works to make it simple to incorporate new component algorithms into the ensemble and is made to run on distributed computer systems. The sites that each algorithm predicts are grouped by the score that each algorithm gives them. Motifs that are in a top group as part of most of the algorithms are identified (all of the predicted sites are given equal weights). The number of times a position in the sequence is identified is counted and a sliding window is used to smooth the score and decide the final site prediction. It works by running the algorithms multiple times, often with different parameters to achieve diverse predictions.
Another ensemble tool, DynaMIT, is completely customizable. It allows users to use whichever motif-finding algorithms, combinations, and visualizations that they choose. In the article, DynaMIT was often tested by running MEME and GLAM2 or Weeder on the sequences, then running the results from each algorithm through novel combining methods, known as integration strategies, such as Jaccard similarity and PCA (principal component analysis) algorithm.
6. Results
As MCAT was first inspired by the study of the cell cycle in fission yeast, we performed an extensive evaluation of MCAT over yeast data sets, including S. cerevisiae and S. pombe. Since S. cerevisiae has been more fully studied compared with S. pombe, we extracted larger data sets from S. cerevisiae. After that, we assessed the performance of MCAT against the well-known comprehensive data sets from the literature (Tompa et al., 2005), which covered DNA sequences from human, fruit fly, mouse, and yeast.
We ran MCAT, EMD, and DynaMIT on these three data sets with specific parameters. We used default settings when executing most of the component tools in MCAT. The default motif width is 8 and the top five motifs were chosen, ranked by comparison score. For MEME and XXmotif, the zero or one occurrence per sequence (ZOOPs) model was selected. As Weeder requires the specification of the organisms, we applied each data set with the corresponding organism. The number of iterations in DECOD is set to 50. We considered only the top five motifs when running the stand-alone tools and ensemble tools for comparison as suggested by Tompa et al. (2005). For DynaMIT, we employed three of the motif search tools in it, MEME, Weeder, and GLAM2. The PCA Strategy was chosen as the integration strategy in DynaMIT.
6.1. Comparison using S. cerevisiae data sets
First, we evaluated MCAT using real S. cerevisiae data sets. These data sets were obtained from Yeastract (Teixeira et al., 2017) (www.yeastract.com). The database contains promoter sequences, documented regulations, TF names with motifs, and corresponding locations. Each promoter sequence has a gene name and sequence. We mapped and joined information into a table, which contains information such as for each sequence which motifs it contains. These will come up with a list of biological upstream sequences (motif is a consensus for a given TF, which is known to regulate a given gene which has this upstream sequence) and biological negative sequences (motif is a consensus for a given TF, which is not known to regulate a given gene that has this upstream sequence). We then picked smaller subsets among those sequences. We used hashing and suffix arrays to manually check if each biological upstream actually contained the motif and marked the upstream as a strong sequence if it actually contained the motif and as weak sequence otherwise. Then, three subsets were generated with different number of combinations of strong sequences and weak sequences. Specifically, the data set Budding Yeast 1 contains 58 test files, Budding Yeast 2 contains 107 test files, and Budding Yeast 3 contains 108 test files. Each test file contains 20 sequences. Sequence is retrieved from up to 1000 bp upstream in the
Figure 5 gives the results of prediction accuracy of running the six tools with three S. cerevisiae data sets. The MCAT-2 designation refers to a variation of MCAT in which the components CMF and Weeder are given 0.4 and 0.6 weights, whereas others have a weight of 1. For MCAT, we selected the top five motifs, ranked by comparison scores, while for EMD and DynaMIT, we took the top five motifs as well. Then we calculated the predication accuracy of the top five motifs from each ensemble tool. The prediction accuracy refers to the binding-site level accuracy, that is, the specificity (sSp). From the figures, we can tell that MCAT improved the accuracy of prediction by about 30% on average compared with EMD. The reason for the low predication accuracy of DynaMIT might be because it does not provide the option to set the width of the target motifs.
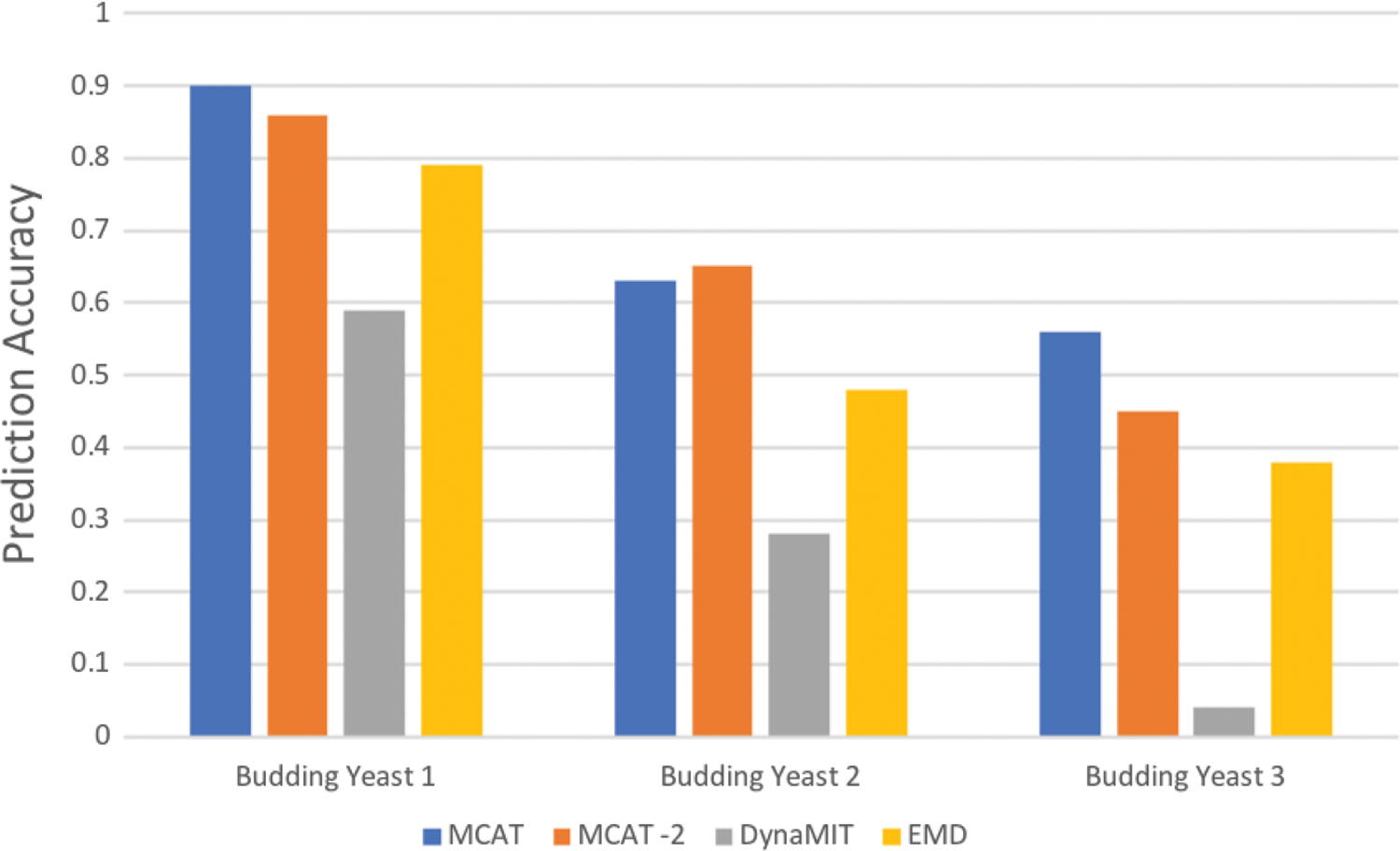
Comparison of Saccharomyces cerevisiae data sets. It displays the prediction accuracy on each of the three DNA data sets. MCAT-2 uses weights for CMF and Weeder that are 0.4 and 0.6, whereas the weights of other tools are 1.
Figure 6 shows the performance of each motif-finding component in MCAT. Compared with Figure 5, the performance of MCAT is better compared with each stand-alone tool. XXmotif gives the best performance compared with other algorithms. However, as users are unable to set the exact number of outputs, XXmotif has a number of false negatives.

Prediction accuracy of each motif-finding algorithm in MCAT. The result shows the performance assessment on three budding yeast Saccharomyces cerevisiae data sets. The y-axis represents the prediction accuracy.
Table 1 shows the range and average running time of each tool with each test file in S. cerevisiae data sets. Figure 7 shows the test on the possible combinations of component algorithms with budding yeast S. cerevisiae data sets and the result of prediction accuracy. Note that the combination of all six tools, BioProspector (BP), MEME (ME), Weeder (WD), DECOD (DE), CMF, and XXmotif (XX), is slightly better than the result of five component algorithms without DECOD, which only improved the result by 5% on average. It was noted that the execution time increases by at least two times in combination with DECOD as the number of iterations increases.
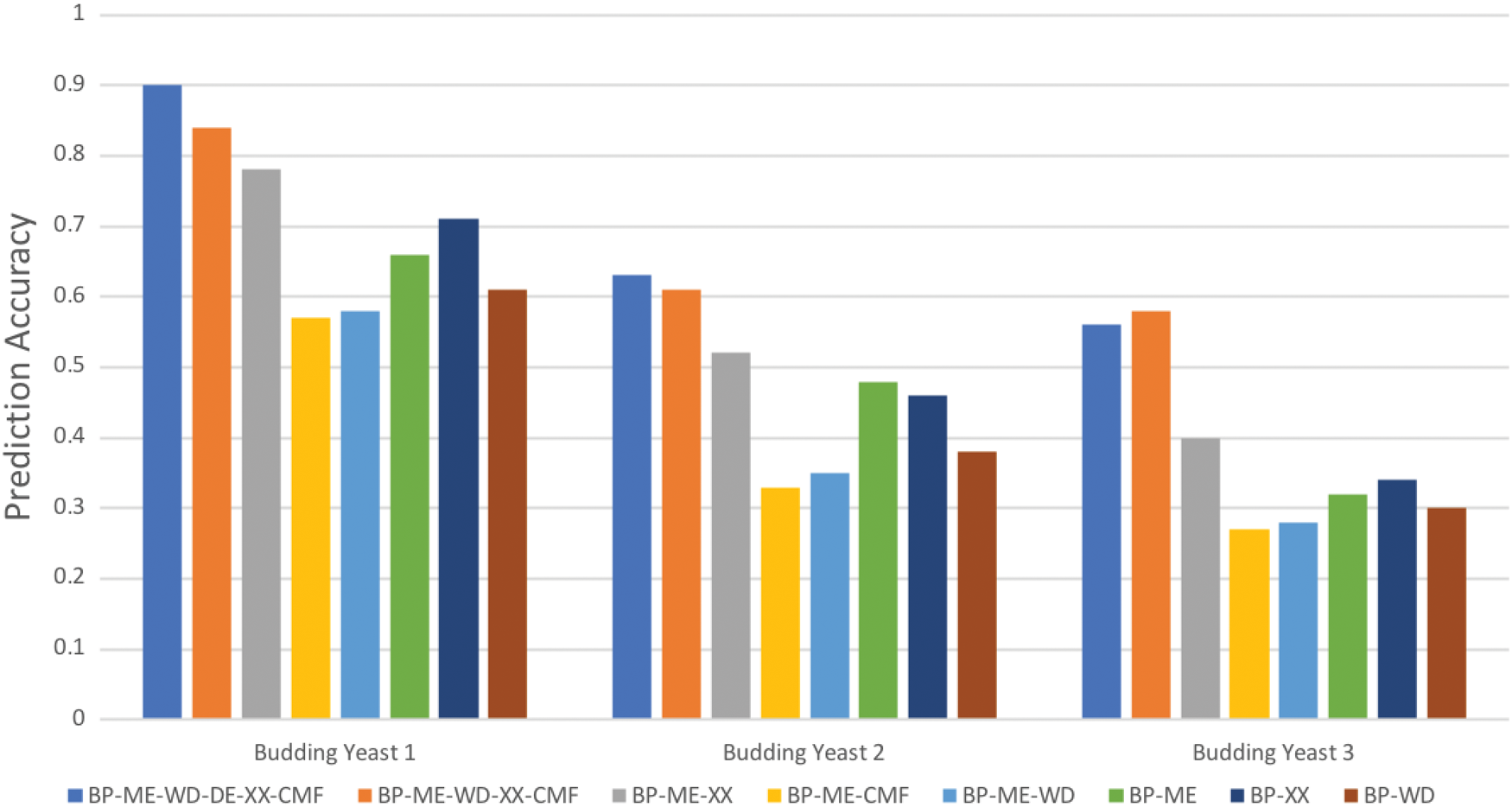
Prediction accuracy of different combinations of component algorithms. BP represents BioProspector, ME represents MEME, WD represents Weeder, XX represents XXmotif. The result shows the MCAT performance assessment on three budding yeast Saccharomyces cerevisiae data sets.
Range and Average Running Time for Finding One Motif in Schizosaccharomyces cerevisiae Data Sets
The values of running time is in seconds. MCAT-3 refers to MCAT with all component tools, except DECOD, and all the tools are equal weight.
MCAT, Motif Combining and Association Tool.
6.2. Comparison using S. pombe data sets
The S. pombe data set for testing was obtained from Oliva et al. (2005) and involves genes related to the cell cycle. In the article, the authors took up to 12,000 bp upstream sequences or to the next upstream open reading frame for motif analysis and applied MEME (Bailey et al., 2009), an expectation-maximization algorithm; AlignACE (Chen et al., 2008), a Gibbs sampling algorithm; and SPEXS (Sequence Pattern EXhaustive Search), a word-count algorithm (Vilo et al., 2000) for motif analysis.
In this study, when testing with MCAT, the data sets we used include a total of 23 genes from the core of the Cdc15 cluster (Cdc15), the 18 genes from the Cdc18 cluster (Cdc18), the 9 genes from the Eng1 cluster, and the 7 genes from the Wos2 cluster. Cdc15 contains five motifs, Cdc18 contains two motifs, and both Eng1 and Wos2 contain one motif. For each gene, the DNA sequence examined was up to 2000 bp upstream of the
Prediction Accuracy of Testing with Schizosaccharomyces pombe Data Sets
Cdc15, Cdc18, Eng1, and Wos2 represent clusters related to the cell cycle. Cdc15 and Cdc18 contain several motifs.
6.3. Comparison using the data set of Tompa et al.
Other data sets used in the performance assessment were from Tompa et al. (2005), which consists of 56 data sets. The data sets consist of real upstream sequences from human, mouse, D. melanogaster, and S. cerevisiae based on the TRANSFAC database (Matys et al., 2003). The original data sets include one benchmark that has the binding sites in their actual genomic promoter sequences; one generic benchmark, which has motifs randomly planted in the promoter sequences from the corresponding organism; and one Markov benchmark, which has motifs planted in sequences generated based on a Markov model. The number of sequences and sequence lengths in these data sets vary; there are between 1 and 35 sequences per data set with sequences of length up to 3000 bp.
Figure 8 shows the performance of individual and ensemble motif-finding tools with the two performance evaluation measures, sensitivity (nSn) and correlation coefficient (nCC). Compared with individual component algorithms MEME, Weeder, and BioProspector, MCAT achieved an average of 320% improvement in sensitivity and 280% in correlation coefficient, whereas compared with ensemble tools EMD and DynaMIT, MCAT gained over 110% in sensitivity and 100% in correlation coefficient.
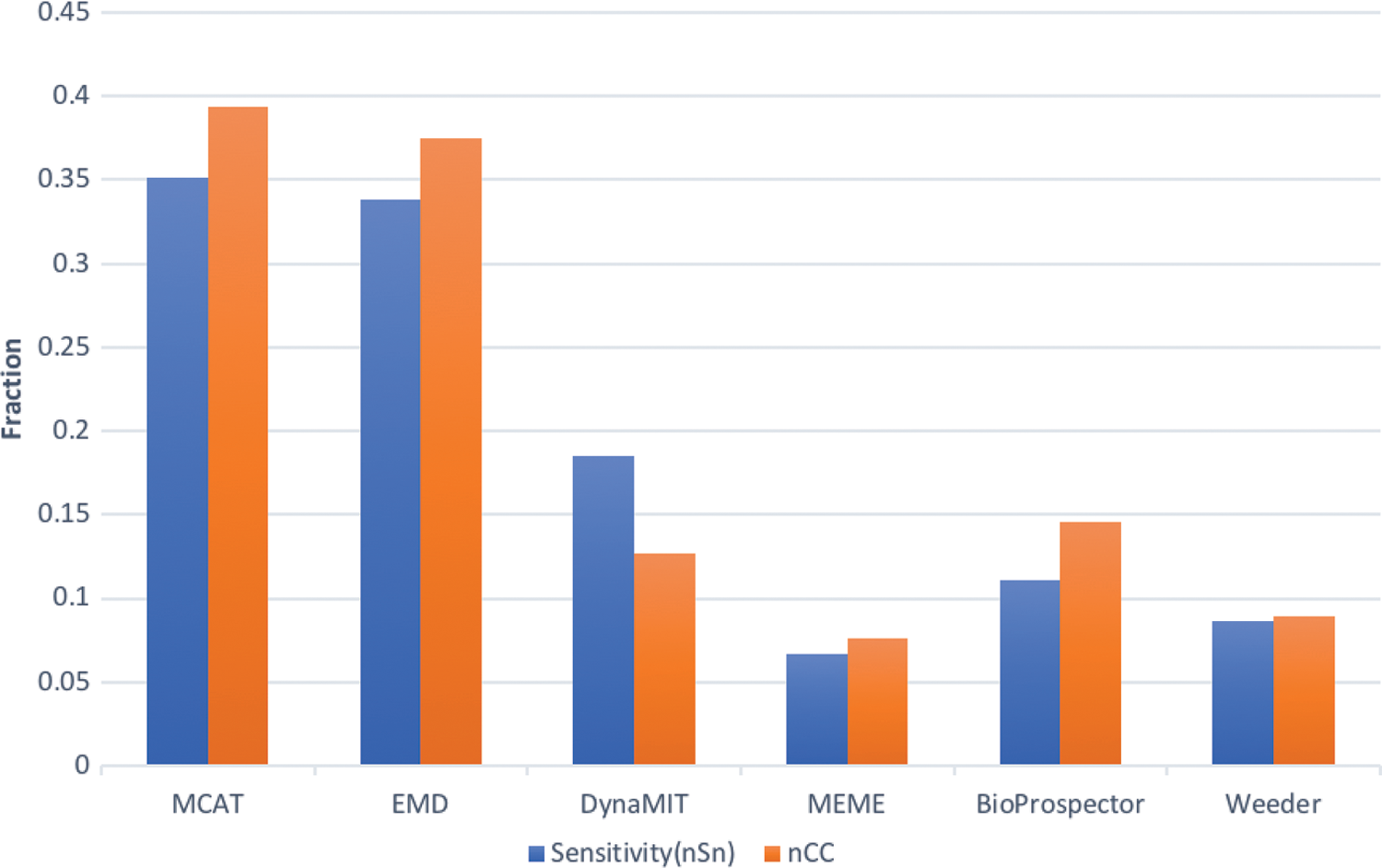
The figure lists the sensitivity (nSN) and correlation coefficient (nCC) of test for Tompa's benchmark. The blue line represents sensitivity and the orange line represents correlation coefficient.
7. Discussion and Conclusion
We developed MCAT, a novel ensemble tool for motif finding in DNA sequences. For developing ensemble motif-finding tools, one key problem is selecting the appropriate component algorithms. MCAT employees six individual motif-finding algorithms, MEME, BioProspector, Weeder, CMF, DECOD, and XXmotif. It achieved good performance according to the results in Section 6. There is no best answer as to the selection of available methods for motif finding; it depends on the features of input data sets and performance evaluation (Schweikert et al., 2012). While different combinations will produce different results, they do improve the results in many specific cases. In MCAT, it is essential that the six tools are diverse. MEME is based on the EM algorithm, BioProspector is based on Gibbs sampling, and Weeder is based on suffix trees. Both CMF and DECOD use a discriminative approach, whereas XXmotif is based on a scoring system with PWMs. These ensure that MCAT has diversity so that motifs with high scores by different algorithms will be more confident and lead to good coverage.
Different combinations of component algorithms lead to different levels of performance on the same data set. Figure 7 documents the prediction accuracy of different combinations of component algorithms. MCAT combines all six tools and has better performance than any individual combinations of tools, as well as in comparison with EMD and DynaMIT. The performance of MCAT with and without DECOD is almost the same. The reason that DECOD does not make a large contribution to the final results might be that the algorithms of CMF and DECOD are highly similar in that both of them are based on a discriminative algorithm. Table 1 reveals that when MCAT employs DECOD, the running time of MCAT increases by more than two times. To obtain a strong result from DECOD, it is necessary to make the number of iterations high, which significantly increases the running time. On the other hand, DynaMIT contains 12 motif-finding algorithms and employs integration strategies for motif finding in DNA and RNA sequences. It allows users to select different algorithms, integration strategies, and result visualizations.
However, it failed to provide clear guidelines for the best combination of tools according to different data sets. In the process of testing DynaMIT, we noticed that some combinations work well while some do not. Another difficulty with DynaMIT is that it lacks global parameters for configuration. Consequently, users are unable to set parameters of target motifs, such as the width of motifs and the number of output motifs, which may cause the low prediction accuracy. A future step is to analyze the quantitative relationship between the diversity of component algorithms and the final results, and provide guidelines for choosing the components of ensemble motif-finding tools when dealing with different data sets.
Another key problem for an ensemble tool is how to combine independent results from different algorithms. The VoteRank algorithm of MCAT ensures that the top motifs have high comparison scores, high PWMs scores, and low pairwise similarity. In the process of combination, we want to make sure the PWMs generated in VoteRank are distinct, as well as the motifs in the final results. Many early motif-finding tools output motifs that are highly similar, which reduces the coverage of the results. The p-value using the QFAST algorithm (Bailey and Gribskov, 1998) and z-score reflect the confidence of each motif. Unlike results from the literature, the p-value in MCAT reveals the combined p-value of one motif against a set of sequences. MCAT used the Markov probability score to find closely related window positions to the query motif. Previous studies (Kim et al., 2014) summarized the ensemble methods, which mainly include naive ensemble methods, scoring functions, and clustering methods. Recently, more machine learning and deep learning methods have been applied on motif-finding problem, such as WSMD (Zhang et al., 2017). Generally, the performance of these methods is better than that of individual motif finders.
To further improve MCAT, identification of appropriate weights of different algorithms and the number of iterations to employee are important issues. In MCAT, we assign weights to different algorithms based on the number of outputs and their performance. According to the result shown in Figure 5, the performance of MCAT with different component weights is not significantly different than those of MCAT with all weights equal to one. One way to optimize the weights is to develop formulas to see whether that improves the performance; another way is to change the number of runs (Hu et al., 2006). However, in MCAT, it is necessary to set an appropriate number of iterations, because BioProspector and DECOD must run iteratively to obtain their final results. If the number of iterations is too low, it is unlikely to guarantee the accuracy of the results.
Overall, MCAT achieves an improved prediction accuracy when compared with individual motif-finding tools and other ensemble approaches. Additional work includes further insights with new data sets and porting to platforms other than Linux, as well as a Web application.
Footnotes
Author Disclosure Statement
The authors declare that no competing financial interests exist.