
Abstract
Multiple transcription factors (TFs) bind to specific sites in the genome and interact among themselves to form the cis-regulatory modules (CRMs). They are essential in modulating the expression of genes, and it is important to study this interplay to understand gene regulation. In the present study, we integrated experimentally identified TF binding sites collected from published studies with computationally predicted TF binding sites to identify Drosophila CRMs. Along with the detection of the previously known CRMs, this approach identified novel protein combinations. We determined high-occupancy target sites, where a large number of TFs bind. Investigating these sites revealed that Giant, Dichaete, and Knirp are highly enriched in these locations. A common TAG team motif was observed at these sites, which might play a role in recruiting other TFs. While comparing the binding sites at distal and proximal promoters, we found that certain regulatory TFs, such as Zelda, were highly enriched in enhancers. Our study has shown that, from the information available concerning the TF binding sites, the real CRMs could be predicted accurately and efficiently. Although we only may claim co-occurrence of these proteins in this study, it may actually point to their interaction (as known interaction proteins typically co-occur together). Such an integrative approach can, therefore, help us to provide a better understanding of the interplay among the factors, even though further experimental verification is required.
1. INTRODUCTION
Multiple transcription factors (TFs) bind to a specific site in the genome. These sites are noncoding DNA sequences of a few hundred base pairs (bp) to several thousand base pairs long and are referred to as cis-regulatory elements (CREs) (Ong and Corces, 2011; Wittkopp and Kalay 2011). The binding of TFs to these CREs may result in the activation of chromatin remodeling (Siggers et al., 2011; Slattery et al., 2011) or the recruitment of a transcription preinitiation complex, which ensures the regulation of genes. The transcription factor binding site (TFBS) motifs are short in length and often imprecise in nature and are referred to as degenerate TFBSs (Stormo, 2000; Bulyk, 2004). This makes the accurate identification of TFBSs an abiding challenge (Wasserman and Sandelin, 2004; Grove and Walhout, 2008). In addition, most of the computationally predicted functional TFBSs are not always available for binding, presumably due to chromatin structure and/or local epigenetic properties. TFBSs often show preferential co-occurrence with other TFBSs (Cao et al., 2010; Yao et al., 2013). The spatial combination of such multiple TFBSs is nonrandom in nature and forms the cis-regulatory modules (CRMs) (Ludwig et al., 1998; Yuh et al., 1998; Krivan and Wasserman, 2001).
For example, even-skipped (eve) stripe 2 enhancer in Drosophila contains multiple binding sites of Kr, Bcd, Gt, and Hb within 500 bp (Small et al., 1992). With the advent of advanced techniques, such as immunoprecipitation of in vivo cross-linked chromatin followed by microarray (ChIP-chip) or sequencing (ChIP-seq), the precise locations of TFBSs can be determined. However, such high-throughput empirical methods remain expensive and technically challenging despite advancements in, and decreasing costs of, the respective technologies. However, computational approaches can be a quicker and more cost-effective solution for CRM or TFBS prediction.
A number of in silico studies have been carried out to determine CRMs (Su et al., 2010; Aerts, 2012; Hardison and Taylor, 2012; Nandi et al., 2013; Suryamohan and Halfon, 2015). These studies (Frith et al., 2001; Pilpel et al., 2001; Berman et al., 2002; Markstein et al., 2002; Rajewsky et al., 2002; Frith et al., 2003; Sharan et al., 2004; Gotea and Ovcharenko, 2008; Kazemian et al., 2013; Spivak and Stormo, 2016) often depend on the computational identification of individual TFBS across the genome. The parameters required for these methods may include prior information on interacting TFs and the expected distance between TFBSs, prior knowledge of the co-regulated genes, and information on specific search areas, such as promoters of certain genes, with a background model for the search space or sequences. Some of the methods (Grundy et al., 1997; Guhathakurta and Stormo, 2001; Sinha et al., 2006; Kim et al., 2010; Wu and Xie, 2010) first deduce the probabilistic grammar rules for the functional module, which are later detected in the genome or preferred search area. Such models predict CRMs by comparing the observed likelihood of the CRMs with the likelihood of CRMs generated by the background models (Aerts, 2012).
As mentioned above, a computational prediction is not free from random co-occurrence, and this could arise from an inappropriate search space. Some methods, instead of considering the whole genome as a search space, target evolutionary conserved space (Chiang et al., 2003; Kellis et al., 2003; Blanchette et al., 2006; Hallikas et al., 2006; Jiang and Singh, 2014; Spivak and Stormo, 2016), which is again not a trivial feat. There are few methods to examine the experimental ChIP sequencing data as a search space to improve CRM prediction (Aguilar and Oliva, 2008). The efficiency of CRM identification can be improved by integrating more information from experimental data. For example, certain regulatory regions, such as enhancers, can be predicted with high accuracy if certain epigenetic features are considered (Hon et al., 2009), such as the monomethylation of lysine 4 of histone H3 (H3K4me1) (Heintzman et al., 2007), the acetylation of histone (Roh et al., 2005; Calo and Wysocka, 2013), or the binding of coactivator CBP/p300 (Heintzman et al., 2007) to the genome.
Several experimental studies have been conducted to identify and categorize the TF-TF interactions in the model organism Drosophila (Giot et al., 2003; Stanyon et al., 2004; Guruharsha et al., 2011; Rhee et al., 2014). A number of studies based on co-expression have been carried out (Tomancak et al., 2007; Adryan and Teichmann, 2010) to explore the TF interactome. Although these studies have categorized a large number of TF-TF interactions in Drosophila, the catalog is still far from complete, since these studies do not include the complete range of TFs. Moreover, there are some databases available that contain a large volume of binding site information of numerous TFs. For example, TRANSFAC (Matys et al., 2006) and JASPAR (Mathelier et al., 2015) contain the binding site information of the TFs in the form of aligned DNA motifs or frequency tables. These databases can be utilized to predict the binding sites and determine the CRMs.
Therefore, to complement the present gap in the experimental studies, we integrated experimental data and computational predictions to obtain additional speculative associations among TFs. This integration helped us to identify novel combinations in addition to the known CRMs. This analysis also identified paired (prd) as an essential factor during developmental processes along with others, such as Zelda (zld). Moreover, after investigating the TF co-occurrence in enhancers, Zelda and Dichaete (D) were found to be highly enriched.
Many different TFBSs can occupy a compact genome locus, referred to as a high-occupancy target (HOT) region (Moorman et al., 2006; Macarthur et al., 2009; Kvon et al., 2012). These studies have revealed that numerous factors, often functionally unrelated, bind to these HOT regions, which result in the activation of nearby genes. Studies in Drosophila (Weigel et al., 1990; Borok et al., 2010) have shown that genomic loci for binding sites of several TFs and cofactors strongly overlap with each other. Previous studies have indicated the sequence signature of certain TFs such as Vielfaltig/Zelda and Trithorax-like/GAGA in these HOT regions to be enriched. In the present study, we identified the HOT regions using data sets similar to those previously used (Consortium, 2010). The observations made in our study indicated that the binding motifs of known TFs are depleted in the HOT regions, although we used more known motifs in our study. Furthermore, the present study has shown that the de novo predicted motifs, Giant, Dichaete, and Knirp, are enriched in the HOT regions.
2. METHODS
2.1. Identification of genomic binding locations/regions of TFs
We determined the genomic binding locations of the recent chromatin immunoprecipitation experiment for the genome-wide binding of 38 TFs in Drosophila (Macarthur et al., 2009; Contrino et al., 2012) by the following method (Supplementary Table S1). All ChIP-chip raw data sets were converted to a current genomic version by using tools available in the UPCSC database (Siepel et al., 2005). The intensity ratio of chromatin immunoprecipitation by antibodies (Ip) and for control DNA input (In) as well as the ratio of Ip to mock precipitation by IgG (IgG) were calculated using the Tiling Array Software (TAS). TAS was used with default parameters and Probe Analysis settings: bandwidth, 300; Test Type, one side upper; perfect match probe intensities. Bound regions were defined by array probes that passed the three standard deviation cutoff within a window size of 200 bp. Regions of less than 5 probes were excluded, and a region was extended as long as there was a value within 200 bp of the previous value. The midposition of the six highest consecutive probe values was set as the center of each detected region. The value of each detected region was set as the average of the highest six probe ratios. After the region had been detected, all the binding values of TFs were linearly adjusted to 0 and 1. In this current study, we compared the identified binding sites with two more available experimental data (Negre et al., 2011; Paris et al., 2013). These studies mapped a large number of TFs to the Drosophila genome. The binding sites obtained from these studies highly overlap with our identified binding sites.
2.2. Prediction of binding sites in Drosophila promoters
The genomic coordinates of the transcription start sites (TSSs) of all known genes were obtained from the Drosophila melanogaster genome (version dm3), downloaded using BioMart (Durinck et al., 2005). The promoter sequences, defined as 1000 bp upstream and 1000 bp downstream sequences around the TSS, were extracted from the Drosophila R5 genome sequence (Marygold et al., 2013). Binding site information in the form of a frequency table was obtained from JASPAR (Version: 5.0 ALPHA) (Mathelier et al., 2015), and position weight matrices (PWMs), which represent the log-odd probabilities of finding each base at each position, were calculated from the frequency table using the method described in Gershenzon et al. (2005) and Nandi and Ioshikhes (2012). The PWMs were then applied to the extracted promoter sequences to predict the binding sites.
The threshold was derived from a background model of randomized promoter sequences, which were created by shuffling the promoter sequences (Nandi et al., 2013). Each PWM was applied to the randomized promoter sequences to build the background model. The background model generated the “Randomized Occurrence Frequency” (OFr) at a given threshold, which essentially represents how many hits could be obtained by chance for the threshold. The threshold value was selected for each PWM by choosing the threshold that generated OFr value of 0.0001 with an intention to minimize the detection of false positives from the promoter sequences.
2.3. Co-occurrence count
In the genomic binding location, if any two factors were found to bind within the range of 100 bp upstream or 100 bp downstream of each other, these factors were considered as associated/co-occurring factors. From the complete genome-wide binding data, the number of times a factor was associated with another factor was retrieved using the criterion. We call this number the co-occurrence count. This co-occurrence count is represented by the thickness of the edges between two nodes, which show the magnitude or number of times each pairwise combination was observed. The widths of the edges were scaled as the ratio of pairwise co-occurrence count of two given factors with the highest pairwise co-occurrence count of any two factors in the particular network. The width was calculated using the following formula:
where μ(a,b) = co-occurrence count of the combination of factors a and b; N = total number of TFBSs.
2.4. Generation of network
The network layout was drawn using Yifan Hu layout implemented in Gephi (Gephi 0.9.1, https://gephi.org) (Hu, 2005). The following parameters were used: optimal distance = 250, relative strength = 0.2, initial step site = 20.0, step ratio = 0.95, adaptive cooling is checked, and theta = 1.2. The bigger the optimal distance and the relative strength make the nodes farther apart. The smaller theta means better accuracy. Eigenvector centrality was calculated with the algorithm implemented in Gephi. The eigenvector centrality is based not only of the degree of the connections to the nodes, but it also favors the connection to the central nodes (Bonacich, 2007).
2.5. Statistical significance of the co-occurring factors
Identified binding sites conforming to the criterion given above were used to determine the co-occurrence. The co-occurrence count was obtained from the designated promoters and compared with the count from shuffled sequences. The z-score was then calculated as follows:
where Obs is the observed co-occurrence count in the designated promoters, and exp is the expected co-occurrence count in the shuffled promoter sequences. The combinations were selected if the z-score is >3.
2.6. Protein–protein interaction
The network data collected from DroPNet (Yu et al., 2008; Renaud et al., 2012) and Cytoscape (Shannon et al., 2003) were used for better visualization and editing. The connections in the network are based on human interologs, DPIM (Drosophila Protein Interaction Mapping) co-AP/MS, or other physical interactions. Human orthologs in Drosophila were used due to the protein interaction data of the corresponding TFs being unavailable. DPIM co-AP/MS (co-affinity purification combined with mass spectrometric analysis) is a Drosophila protein interaction map using affinity-tag-based protein purification followed by mass spectrometry-based proteomics. Other physical interaction data sets are based on a yeast two-hybrid system retrieved from DroID (Renaud et al., 2012).
2.7. Identification of the HOT regions
The binding profiles of 38 early embryonic TFs were used to identify the genomic regions where more than one TFBSs were found within 200 bp. The central peak of a TF binding region was used as an initial point, and all other peak centers were included within a certain region if their peaks were not separated by more than 200 bp from each other. The HOTness value of a genomic region was calculated based on the number of TFBSs within the genomic region. For the occurrence of each TFBS in the region, 1 was added to the HOTness value. And if the TFBS was already present in the given region, 0.5 was added to the HOTness value (Supplementary Fig. S1). All the determined TFBSs were assigned to one of five classes according to their HOTness values. Class I contained TFBSs with a HOTness value of 1 to 1.5, Class II (2 ≤ HOTness <6), Class III (6 ≤ HOTness <8), Class IV (8 ≤ HOTness <13), and Class V (13 ≤ HOTness). Regions in which ≥8 TFs bind were defined as the HOT regions.
2.8. De novo motif identification
We used previously developed tools, Sigfinder [Sigfinder V2012, with default parameters with probe setting parameter: createProbesBylength(6)] and Aligner (Aligner V2012, with default parameters), for de novo motif discovery (Philip et al., 2012). Supervised multivariate method, orthogonal partial least squares discriminant analysis (OPLS-DA) (Bylesjo et al., 2006), was implemented to identify motifs or sequences that could be used to predict TF recruitment in the HOT regions. For motif prediction, the peak centers were used together with the region, which extended to 100 bp both upstream and downstream. Only intronic regions of Class I HOTness that were strongly bound by the corresponding TFs were selected as a positive set. Class I regions, which were bound by TFs other than their corresponding one, were randomly selected and used as a training set for constructing the model. The top 1000 sequence words from each OPLS-DA model were used. Aligner detects sequence differences between genes strongly bound and weakly bound by the TFs based on the top 1000 sequence words obtained from the OPLS-DA models.
2.9. Identification of enhancers
Heintzman et al. (2007) have shown that enhancers are enriched with H3K4me1 and CBP/p300 protein, which form the signature of active transcriptional enhancers. In the present study, to predict the enhancers, the H3K4me1 regions and CBP/p300 bound regions were obtained from modENCODE (Consortium, 2010) for different time points, from early stage to 12 hours. These regions were then overlapped, which narrowed down the search space.
3. RESULTS
3.1. Higher association and CRMs among the TFs at early developmental stages
The experimentally determined TFBS information (Macarthur et al., 2009; Contrino et al., 2012) was used to determine the cooperative binding or association among the factors across the genome. Based on the developmental stages considered, the data were divided into two groups: 0–3 and 0–12 hours. From the experimentally identified genomic binding locations, closely associated or co-occurring TFs were used to derive a network (Fig. 1A). Factors Vielfaltig/Zelda (zld), Ultrabithorax (Ubx), Zn finger homeodomain 1 (zfh1), twist (twi), and Stat92E (stat) were observed to be highly connected with all the factors in the experimental study. These factors were followed by snail (sna), schnurri (shn), zeste (z), paired (prd), sloppy paired 1 (slp), runt (run), medea (med), and Mothers against dpp (Mad). Previous studies have indicated that these factors are involved in the developmental processes (Lewis et al., 2000; Castanon et al., 2001; Czermin et al., 2002; Leatherman and Dinardo, 2008; Liang et al., 2008). The observation of high connectedness of these factors in the network indicates that their co-occurrence is essential for the process of development.

TF-TF interactions derived from experimental data (Giot et al., 2003; Adryan and Teichmann, 2010).
The number of factors in the 0–3 hours (Fig. 1B) and 0–12 hours (Fig. 1C) data sets were 23 and 15, respectively. The factors present at 0–3 hours (Fig. 1B) seem to associate very strongly as presented in the thickness of the connections in the overall interaction (Fig. 1A) and can be observed as the factors toward the center of the network. Investigating the number of co-occurrence of each factor with others revealed that the factor prd was connected with other 21 factors. The mutual positioning of the other factors in the whole genome with respect to prd was investigated to determine if there were any positional preferences in terms of their distances from each other (Fig. 1D). Figure 1D reveals that prd combines with other factors in an overlapping manner, for example, Mothers against dpp (Mad), twist (twi), daughterless (da), and so on, which is shown using blue dots in the figure. The spread of these factors with respect to the prd binding site was very restricted and is represented by the red bars in Figure 1D. The second most highly connected factor was zld in the 0–3 hours network (Fig. 1E). This analysis shows that the ChIP data used in the analysis were of good resolution. This resolution can be understood from the distribution of the factors with respect to prd and zld in Figure 1D and E, respectively. The distribution represented by the red bar shows that the spread is confined.
In the 0–3 hours data, a previously reported daughterless-twist (da-twi) (Wong et al., 2008) was observed as the highest co-occurring factors. Following the pair, dorsal (dl) was found to be paired with several other common factors, including Zelda, Twist, Dichaete, and Daughterless. The co-occurrence of many factors with Dorsal signifies Dorsal as being an important factor during the early developmental stage of Drosophila. This pair was followed by many factors paired with Ultrabithorax (Ubx). The factors paired with Ubx were as follows: bab, cnc, GAGA factor (GAF), and Distal-less (Dll). However, the most highly interacting factors were zld, twi, and z in the 0–3 hours network; and zfh and ubx in the 0–12 hours network. Previous studies show that these are essential developmental factors. This study also reveals the importance of these factors from the binding sites association investigation.
3.2. Predicted TFBS associations
Co-occurrence of factors from JASPAR database (Mathelier et al., 2015) was identified following the criteria used in the previous section (Fig. 2A). The connections represent the pairwise combination of two factors in promoters. The width of the edge between two factors represents the count of pairs in the promoters. In the network, mirr was found to be the most highly connected. This high interaction, however, was due to the short and low complexity motif of mirr, which increased the possibility of discovering a large number of random hits. Mirr was therefore removed from the downstream analysis to avoid the noise.
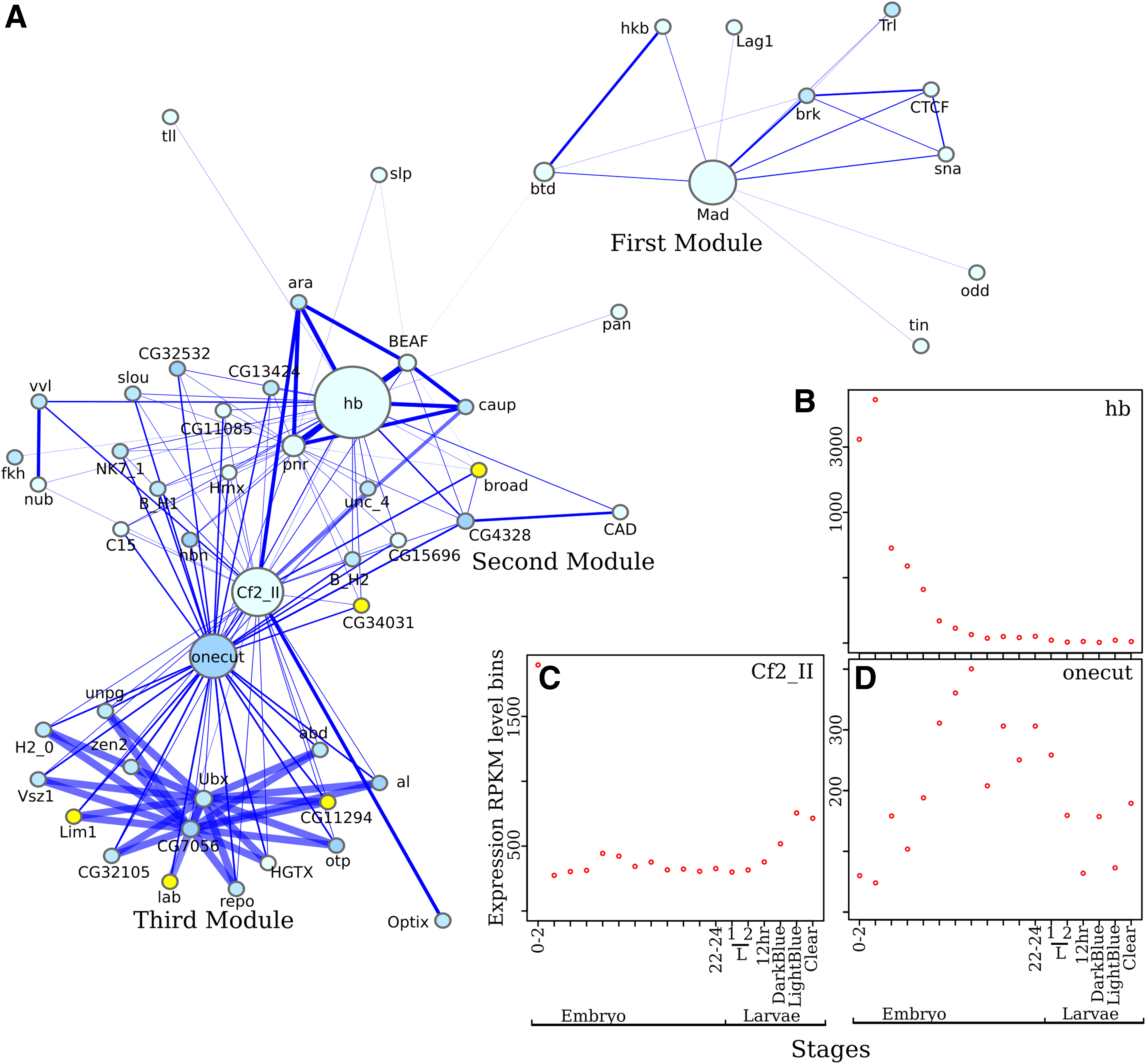
TF interaction network generated from predicted TFBSs.
The obtained network was found to have three distinct clusters or modules; these were termed as the first, second, and third module. The expression pattern of all the factors along different developmental stages of Drosophila was integrated into the network (Graveley et al., 2010). The nodes of the network were colored to show the time point at which the expression of TFs reaches the peak: 0–6 hours, 7–12 hours, and later than 12 hours (Fig. 2A). Interestingly, the first module was popularized by the factors with peak expression during the 0–6 hours of development. The second module was populated with both 0–6 and 7–12 hour factors, and the third module was populated with factors of later than 12 hours of development.
Until recently, of around 700 TFs, only 38 were experimentally mapped to the Drosophila genome. To determine whether there were any other possible associations of factors not otherwise included in the ChIP experiments (Contrino et al., 2012), the ChIP experiment was overlapped with the predicted co-occurrence network from JASPAR. Common factors between the two networks were selected, and a subnetwork was generated from these selected factors as shown in Figure 3. The green nodes in the network indicate those factors common between the two networks, whereas the orange ones are the factors only identified in one of the networks. Essentially, this analysis highlighted the additional associations with the factors in JASPAR, which were otherwise missing from the experimental data. The edge widths of the networks were also adjusted based on the co-occurrence count. From this analysis, we identified factors with strong co-occurrence counts, such as Mother against dpp (Mad)–snail (sna)–brinker (brk)–CTCF, huckebein (hkb)–Mad–Buttonhead (btd), caudal (CAD)–CG4328–hunchback (hb), and Homeobox protein caupolican (caup)–hunchback (hb)–Homeobox protein araucan (ara). These factors individually have a role in chromatin organization and developmental processes. However, their roles in combinations need to be explored.

Overlapping network. The network generated with the common TFs between the ChIP experiment and the JASPAR database.
3.3. CRMs in enhancers
Previous studies and comparative genomics have revealed that regulatory elements involved in early eukaryotic development are located at remote sites, far away from the TSS of their target genes (Weigel et al., 1990; Lettice et al., 2003; Nobrega et al., 2003; Woolfe et al., 2005; Borok et al., 2010). In this study, the binding sites were categorized as proximal and distal according to their position with respect to the TSS and the enhancers. If the distance of the binding sites were ≤1000 bp to the TSS than we regarded them as proximal and if the distance of the binding sites were >1000 bp from TSS, we considered them as distal/enhancers. The networks were generated by connecting two TFs if their binding sites are present close to each other within ±100 bp (Supplementary Figs. S2 and S3). We observed around 15,000 and 36,000 sites in the proximal and distal regions, respectively. The network generated from these segregated data revealed that Ultrabithorax (Ubx), Twist (twi), Dorsal (dl), and dachs (d) were highly co-occurring factors in the proximal region. However, Ubx was not detected to be interacting in this manner at enhancers. In enhancers, Runt (run), Zelda (zld), Invected (inv), Sloppy paired 1 (slp1), and Giant (gt) were found to be highly connected with other factors. This difference indicates that these factors exhibit a preferential binding pattern with respect to the TSS.
3.4. Genome-wide identification of the HOT sites
Some recent studies have shown the presence of large numbers of TFBSs at the regulatory regions, termed as the HOT regions, and large numbers of single factor binding sites available in the HOT regions in Drosophila have been demonstrated (Bylesjo et al., 2006; Roy et al., 2010). The binding profiles of 38 early embryonic TFs were used to identify 21,146 genomic regions to which between 1 and up to 22 TFs were bound within 200 bp of each other. The HOT regions were scored or a HOTness value was assigned to each HOT regions based on the occurrence of the TFBS. A total of 1244 regions were found to be the HOT regions, which comprised 5% of all binding sites. However, most binding sites were single factor binding sites or Class I binding sites, which comprised 67% of the total (Supplementary Fig. S4).
3.5. Known motifs of TFs are depleted in the HOT regions
The known motifs of the TFs in the different HOTness class regions were examined to determine whether the recruitment of factors to the HOT regions was specific. The enrichment scores of known regulatory motifs of the 30 TFs were calculated throughout all TFBSs. To determine the enrichment score, we have used an in-house built algorithm written in Perl where we searched the PWM around a genomic region in bins. The length normalization was carried out to optimize the variation in genomic regions. The motif score cutoff was set based on the average enrichment score in a similar number of the randomly selected control region from the genome of variable length. The motif enrichment was calculated using the hypergeometric test by comparing with the probability of observing the given number (or more) of target sequences with the motif by chance. The aim was to observe if the presence of individual TFs could be predicted using these motifs. Most of the TFs were commonly present in the HOT regions while their DNA motifs were not (e.g., Hairy and Snail). Other factors, such as GAF, Dorsal, and Bicoid, seemed to correlate well with their binding motifs. These differences are not likely to be caused by differences in motif quality since most motifs correlate well with the TFs presence at the sites of low HOTness. It is well known that many TFs interact physically, and it may be true that some are mainly recruited to the HOT regions by other TFs.
3.6. HOT regions have a specific sequence signature
Thirteen TFs were selected based on their enrichment in the high HOTness HOT (Fig. 4, Clusters 1 and 2). These 13 TFs were used to identify the motif enrichment in the HOT regions. After the de novo motif discovery for all the 13 TFs, both the known motifs and the predicted motifs were mapped across the peak centers of each TFBS. In most cases, motifs of the selected TFs were not enriched in the HOT regions, whereas the presence of some TFs' motifs decreased with increasing HOTness. Although some TFs were enriched in the HOT region, their dependency on motifs to bind in the HOT regions disappears. However, three de novo motifs of TFs (Giant, Dichaete, and Knirp) were enriched in the HOT regions. Their predicted motifs were similar, and motifs of these three factors reported earlier do not seem to be present in their binding sites. De novo motif discovery in the HOT regions revealed an enrichment of a common motif CAGGtA in both promoter and intronic HOT regions (Supplementary Fig. S5). This motif is very similar to the zld motif. Other predicted motifs do not match with any other known motifs of TFs of D. melanogaster.

TF occupancy among different classes of HOTness. Occupancy (in percentage) is calculated according to the presence of the corresponding TF against the total number of TFBS within a particular class. TF occupancy is calculated according to their presence as a percentage in a particular class. For the 13 TFs (Clusters 1 and 2), the occupancy increases with increasing HOTness value. Clusters 1 and 2 represent the HOT sites. K-means clustering was used to group the 13 TFs into 4 clusters. HOT, high-occupancy target.
4. DISCUSSION
The CREs in genome contain a set of TFBSs. The nonrandom binding of TFs or CRMs to the CREs determines the transcriptional regulatory activity of them. Therefore, identifying and characterizing these modules is highly essential to understand the gene regulation mechanism. The genome of Drosophila contains around 14,000 genes, and ∼708 genes are TFs (Hammonds et al., 2013; Rhee et al., 2014). However, to the best of our knowledge, the experimentally determined genome-wide binding information is available only for the 38 TFs (Macarthur et al., 2009; Contrino et al., 2012). Therefore, the associations among other factors have not been determined. The factors examined in these experimental studies were mostly developmental factors. The studies investigated the binding site association of these 38 factors and disclosed that some of these factors are associated strongly with other factors, such as zld, z, ubx, and zfh. This strong association may indicate that these factors play an important role in a concerted manner during development. To compensate the limit of 38 experimentally studied factors, computational prediction of the binding sites was therefore integrated with experimental data to compensate for this gap in our knowledge.
The primary objective of this study was to identify the novel association and interactions of the TFBSs. This integrative analysis identified novel associations, such as Mad-sna-brk-CTCF, hkb-Mad-btd, pnr-slp1-BEAF_32, and CAD-CG4328-hb. From the gene ontologies from FlyBase (flybase.org) (Gramates et al., 2017), it is evident that these factors play a role in development. Therefore, it is crucial to study these associations and determine the role they play together. In addition, CRMs reported in previous studies were also detected in our study. For example, factors defining the anterior–posterior axis of the embryo Bicoid (bcd), Caudal (cad), hunchback (hb), Kruppel (kr), and Knirps (kni) were detected. Factors participating in development and differentiation, such as repo, zen, unpg, and Lim1, were also observed as CRMs. Apart from the integrative analysis, the genome-wide binding profile of 38 TFs in Drosophila was exploited to determine the associations among them.
In addition to the identification of the existing associations such as daughterless-twist (da-twi), we discovered novel associations such as between kn and zfh. These factors are individually involved in the formation and development of somatic muscle and wing pattern formation (De Celis, 2003; Leatherman and Dinardo, 2008). Involvement of this combination in any biological or developmental activity of Drosophila was not found in the curated Drosophila transcriptional CRM database, “REDfly” (v5.2.2) (Gallo et al., 2011). From FlyBase (Gramates et al., 2017), however, it was found that kn and zfh take part in the developmental process independently. It needs to be established, what function they perform being together. Some genes, such as Lamp1, sxc, Eph, Hand, and CalpA, were found to have this combination in their proximal promoter, and FlyBase reveals that these genes are involved in the developmental processes of Drosophila. Moreover, expression data reveal that these genes are expressed during the early stages of development. This observation suggests that the novel combination, kn-zfh, may regulate the expression of these genes. We do not yet know what function they perform in combination. Other enriched pairs of TFs were also observed with Ubx. Marin et al. (2012) demonstrated that Ubx plays a critical role in conferring segment-appropriate morphology. The factors paired with Ubx were as follows: bab, cnc, GAF, and Distal-less (Dll).
Previous studies have revealed that these factors are involved in the developmental processes, including limb formation, labral and mandibular development, and pattern formation (Kopp et al., 2000; Lewis et al., 2000; Kojima, 2004; Coulcher and Telford, 2012). Since these factors individually are involved in the developmental process, they might cooperate with each other to accomplish their function, and their associations should be studied further to investigate their combinatorial role in development. The co-occurrence of central regulatory factors, such as dorsal or zld, was also observed in the early hours of development but not in later stages. Dorsal was found to be paired with a number of other common factors, including Zelda, Twist, Dichaete, and Daughterless. Dorsal is also known from previous studies as the focal protein for dorsoventral polarity in developing Drosophila fly (Roth et al., 1989; Rushlow et al., 1989; Steward, 1989; von Ohlen and Doe, 2000). The observation of Dorsal as the central factor in the present study corroborates these other findings concerning its importance during development.
In the present study, the co-occurrence of the factors was investigated in the enhancers. It was found that Zelda remained as the most connected factor. Factors co-occurring with Zelda are known for their involvement in development. For example, Dichaete is involved in central nervous system (CNS) development in Drosophila (Ma et al., 2000; Overton et al., 2002), and daughterless is involved in sex determination and neural differentiation (Vaessin et al., 1994; Zeitlinger et al., 2007). The higher occurrence of Zelda was followed by Dichaete or Sox Box protein of high-mobility group in enhancers. Previous studies have shown that this factor, along with enhancers-binding protein, is capable of binding and bending DNA (Travis et al., 1991). Enhancers are also known to be able to regulate multiple genes (Mohrs et al., 2001; Pennacchio et al., 2013). Furthermore, from a previous study (Liang et al., 2008), it is clear that Zelda initiates the transcriptional activation during the initial stages of development. The observed positioning of Zelda and Dichaete binding sites at enhancers may, therefore, be strategically necessary for the initiation of transcriptional activation of a vast number of genes during the early stage of development. This observation corroborates the findings of previous work that regulatory elements involved in early eukaryotic development are located at remote sites, far away from the TSS of the target genes (Weigel et al., 1990; Lettice et al., 2003; Nobrega et al., 2003; Woolfe et al., 2005; Borok et al., 2010).
Investigating the number of co-occurrence of each factor with others in the network (Fig. 1A) revealed that the factors prd and zld were most highly connected factors. However, little information regarding the interaction of prd with other TFs has been reported in the literature to date. Figure 1D and E reveals that these factors combine with other factors in an overlapping manner. Higher connections with zld were expected, as a previous study using global expression profiling confirmed that zld has a major role in the activation of the early zygotic genome and suggested that zld may also regulate maternal RNA degradation during the maternal-to-zygotic transition (Liang et al., 2008).
By using these 38 TFBSs from the experimental data, we were able to determine a large number of HOT sites. It was observed that the motifs of known TFs were depleted in these HOT regions. The absence of a known motif was not due to the low quality of the motif because most of the motifs correlated well with a single factor binding site or region with a lower HOTness value. Therefore, to understand the phenomenon, we searched for a de novo motif for each TF among the HOT sites and single binding sites of the corresponding TF. De novo motif predictions also exhibit the same phenomenon. This indicates that the binding of TFs to HOT sites is a more complex process than being merely by sequence specificity. Binding of one TF to the genome may recruit additional TFs in the neighborhood through protein–protein interactions. TFs in the HOT sites tend to be highly interactive with other TFs in the protein–protein interaction network (Fig. 5).

Protein–protein interaction network. Protein interaction networks based on human interologs data collected from DroPNet (Yu et al., 2008; Renaud et al., 2012) and Cytoscape (Shannon et al., 2003) were generated by using the information for known interactions of human proteins to show the interaction of their orthologs in Drosophila.
For example, Mad, Ubx, dl, and bcd all seem to be highly interactive with other TFs; this was also shown in the computationally generated network (Figs. 1A and 2A). Furthermore, to determine the missing interactions in the integrated experimentally and computationally generated networks, we explored protein–protein interaction networks. In doing so, we found that twist (twi) and daughterless (da), as well as knirps (kni) and Kruppel (Kr), were involved in the protein–protein interaction. The protein–protein interaction network also suggests the presence of intermediary TFs that might help to recruit other TFs to the HOT sites by protein–protein interactions. More experimental data would provide us with the true structure and function of TF networks and their role in development. The HOT regions were also found to be enriched with the Giant, Dichaete, and Knirp binding motifs. A common TAG team motif, CAGGtA, was observed at both promoters and introns HOT regions, which might play a role in recruiting other TFs.
5. CONCLUSIONS
Our results were drawn from the resources available including the information of the TFBS, which has yet to be completed. The efficiency of such an approach depends primarily on the data generated from experimental studies. However, by integrating the existing resources with the ChIP sequencing data and the computational prediction of TFBSs, we have been able to identify novel combinations. These hypothetical CRMs so discovered should be studied further to determine their role in the development of Drosophila. Although we only may claim co-occurrence of proteins in this study, it may actually point to their interaction (as known interaction proteins typically co-occur together). To check the latter hypothesis, experimental verification is required. As more information becomes available in the future, more precise predictions will be possible. Such enriched information could then lead to a better understanding of developmental processes and improve our knowledge of any interactions involved. Such an approach would also be of great benefit in elucidating the interacting pairs of factors involved in diseases. Although we discovered novel interactions in this study, further study will be required to establish the full range of functions these factors perform when they operate cooperatively.
Footnotes
ACKNOWLEDGMENT
We thank Dr. Mads Kaern for critically reading the article, providing helpful comments, editing, and proofreading the article.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
S.N. was supported by a Ramalingaswami Fellowship (BT/RLF/Re-entry/48/2013) from the Department of Biotechnology, Government of India. The funding body had no role in the design of the study, data collection, analysis, interpretation of data, writing of the article, or the decision to publish.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.