
Abstract
Abstract
Mining meaningful and comprehensive molecular characterization of cancers from The Cancer Genome Atlas (TCGA) data has become a bioinformatics bottleneck. Meanwhile, recent progress in cancer analysis shows that multi-omics data can effectively and systematically detect the cancer-related genes at all levels. In this study, we propose an improved particle swarm optimization with dynamic scale-free network, named DSFPSO, to extract features on multi-omics data. The highlights of DSFPSO are taking the dynamic scale-free network as its population structure and diverse velocity updating strategies for fully considering the heterogeneity of particles and their neighbors. Experiments of DSFPSO and its comparison with several state-of-the-art feature extraction approaches are performed on two public data sets from TCGA. Results show that DSFPSO can extract genes associated with cancers effectively.
1. Introduction
As cancers seriously endanger human health, it is necessary to find the underlying mechanism of cancers to provide a solid foundation for disease prevention, diagnosis, individualized therapy, and drug development. Along with the rapid development of microarray and high-throughput sequencing technology, a large biological data of different categories have been generated. The Cancer Genome Atlas (TCGA) presents multiple types of cancer at different levels, which can help us to learn the biological mechanism of cancers (Bersanelli et al., 2016).
However, most studies carried out based on TCGA mainly focus on a single category of genetic data. Since the development of cancer is a chain event, the single level of omics data cannot fully illuminate the underlying mechanism of cancers. The multi-omics data can effectively remove random events and systematically detect the cancer-related genes at all levels, which would provide innovative perspectives on disease risk assessment, personalized treatment, and medicine guide. So it is acknowledged that integrating these heterogeneous data would present more biological insights than performing separate analyses. Nevertheless, copy number, mutation, and DNA methylation data are closely linked in biology, but different widely in data structure, which makes it difficult to integrate. So it is a bottleneck to integrate single levels of omics data and mine meaningful and comprehensive molecular characterization of cancers from TCGA data.
Among the many ways to illuminate underlying mechanism of cancers from TCGA, extracting features is an effective way. The availability of large quantities of biological data has enabled a variety of feature extraction methods to achieve impressive breakthroughs, such as least absolute shrinkage and selection operator (LASSO), penalized matrix decomposition (PMD), and sparse principal component analysis (SPCA). Roth (2004) used the generalized LASSO method to feature selection problems for microarray data. Liu et al. (2014b) carried out differential expression analysis on RNA-seq count data based on PMD. Luss and D'Aspremont (2010) applied SPCA for clustering and feature selection. Although LASSO, PMD, and SPCA have achieved satisfactory performance on explaining the biological data, current feature extraction methods struggle in tackling problems on multi-omics feature analysis. Specifically, these conventional feature extraction methods excel at identifying genomic feature from single type of genomic feature, but do not offer a satisfactory solution for the integrated multi-omics data analysis. This significant gap between the single level of omics data and multi-omics data analysis creates fertile ground for feature extraction developments.
For this reason, we consider a simple way of improving feature extraction method by leveraging particle swarm optimization (PSO). Recently, many PSO-based methods have been proposed for determining SNP–SNP interactions (Zhang et al., 2016), gene features selection (Chuang et al., 2008), and cancer classifications (Kar et al., 2015). PSO is a population-based search algorithm of adaptive evolution, which was proposed by Kennedy and Eberhart (1995). Owing to its simple structure and fast convergence, PSO has become an important evolutionary algorithm. In recent years, numerous studies have been carried out to improve the performance of PSO. Kennedy and Mendes (2002) have conducted a deep research on population structure and particle behavior, finding that topology has a profound impact on particle behavior. Liu et al. (2014a) proposed SF-PSO (scale-free PSO) that incorporates scale-free topology to improve the optimization process with respect to its solution quality and convergence velocity. Gao et al. (2015) proposed SIPSO (selectively informed PSO), which employs scale-free network to simulate the population structure and selects different learning strategies based on particle degrees. The DMS-PSO uses random dynamic changed population structure that greatly improves the ability of local search (Zhao et al., 2008).
However, conventional improvements on PSO algorithm suffer from the limitation of particle population structure. For example, SF-PSO and SIPSO generate the population structure before experiments, which cannot embody the dynamic changes in the process of iteration. DMS-PSO achieves the dynamic changes in population structure to a certain extent, but the population structure building becomes a completely random process, which is unable to fit in with the actual optimization problems.
To address the aforementioned two issues in conventional extraction methods and the existing improvements on PSO algorithm, we change the conventional particle population structure and propose an improved PSO-based algorithm with dynamic scale-free network, named DSFPSO, to detect multi-omics features.
The contributions of this work are highlighted as follows:
The introduced scale-free network as the particle population structure can be dynamically adjusted in the process of iteration. Four types of velocity updating strategies are used in DSFPSO for fully considering the heterogeneity of particles and the connecting between neighbors. To utilize the difference at different levels of multi-omics data, we resort scoring scheme to extract genomic features that gives priority to genes with top ranks or multiple ranks. We carry out integrated analysis on multi-omics data. To evaluate the effectiveness of DSFPSO, we performed experiments on two data sets provided by TCGA and compared the results with other methods. The identified genes are appraised by gene function analysis and pathway analysis. Extensive experiments demonstrate that DSFPSO can identify disease-associated features effectively.
The remainder of the article is organized as follows. Section 2 describes the proposed method. Section 3 elaborates the experimental results on TCGA database. We compare DSFPSO with other feature extraction methods in this section. In Section 4 we conclude this article.
2. Methods
2.1. Basic PSO algorithm
PSO is similar to other evolutionary algorithms that use the concepts of “groups” and “evolution.” The basic idea of this algorithm is that each individual can be regarded as a particle without bulk and density but with a certain velocity in D-dimensional search space (Eberhart and Shi, 2002). The velocity of each particle can be dynamically adjusted according to the particle itself and its peers' experience based on the fitness value. Based on the fitness value of the position, each particle will move to a better place and obtain the optimal solution of optimization problems.
The basic PSO algorithm can be illustrated as follows:
Step 1: Initialize the particle velocity and position.
Step 2: Evaluate the fitness of each particle.
Step 3: Decide whether to update personal and group best positions by comparing the fitness value.
Step 4: Update the position and velocity of the particles.
Step 5: If the end condition is not satisfied, return to Step 2.
The particle swarm is initialized into a group of random particles, and then the optimal solution is found by iteration. At each iteration, each particle updates the location of the particle by tracking its previous optimal location and the optimal location of all particles. The evolution equation of basic PSO is shown in Equations (1) and (2).
where
2.2. DSFPSO on multi-omics data
In this study, we propose DSFPSO with dynamic scale-free network as its tailored population structure to mitigate the issue in the conventional improvements of PSO algorithms, and capitalize DSFPSO on multi-omics data analysis to extract features. The flowchart of the proposed method is shown in Figure 1. We describe DSFPSO in detail from six aspects.
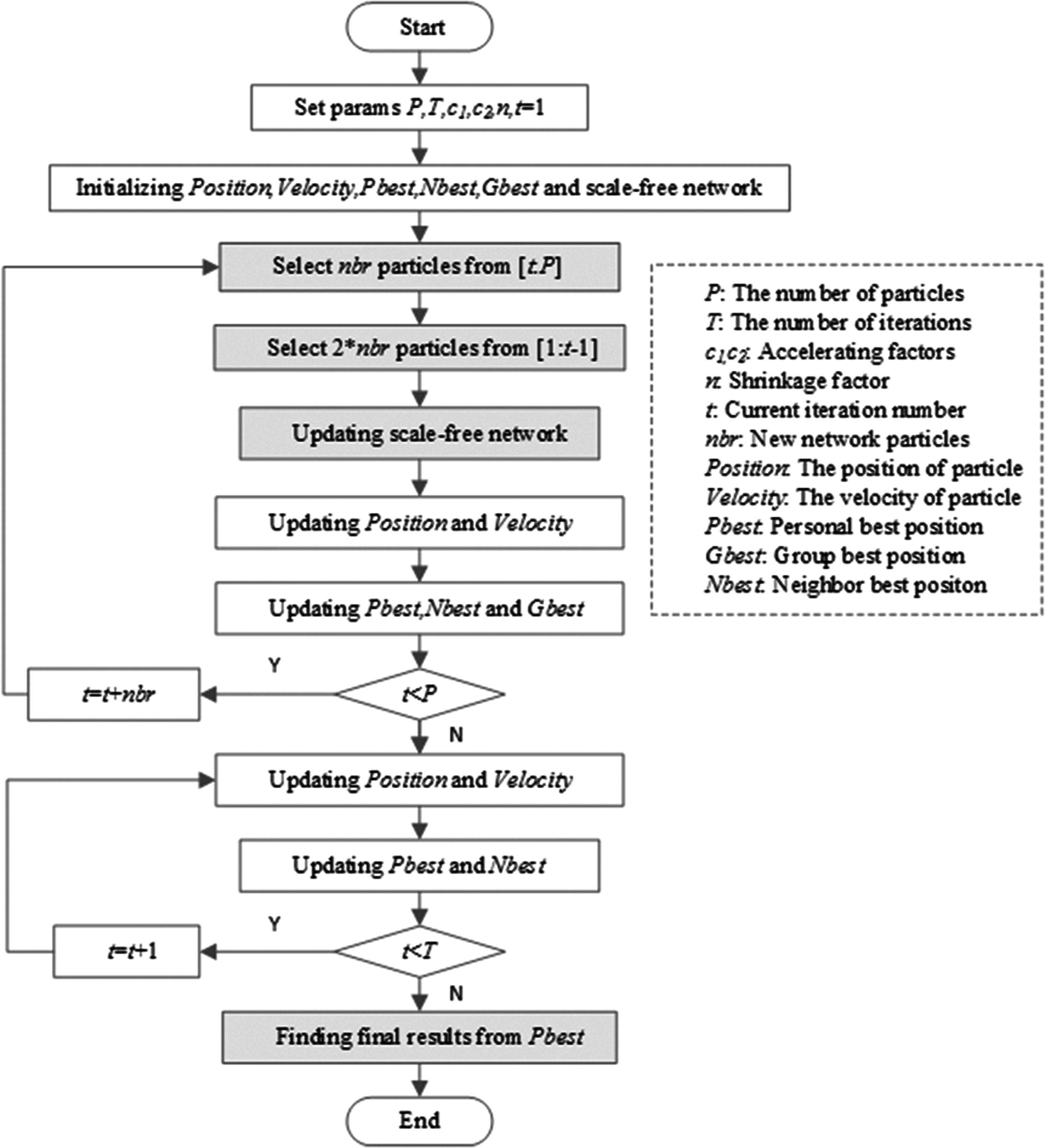
The flowchart of DSFPSO and the components with blue background are the highlights of DSFPSO. DSFPSO, particle swarm optimization-based algorithm with dynamic scale-free network.
2.2.1. Initializing particles with multi-omics data
According to the characteristics of the omics data, we integrate the data as genomics and clinical information matrices. The whole genomics matrix is the search space of particles, whereas the clinical information matrix is used for the test of particle fitness value.
Based on the mentioned mapping of multi-omics data, the position of particle i at iteration t can be illustrated as
The velocity of particle i at iteration t can be defined as
Similarly, the personal best position, neighbor best position, and group best position of particle i at iteration t can be defined as
2.2.2. Analysis of the fitness function
In DSPFSO, fitness function plays a vital role on deciding which genomic feature is the disease-related feature and measuring how much the effect of a captured genomic feature to the phenotype is. Since mutual information does not need to assume the distribution of genomics data and can effectively measure the nonlinear relationship between genetic characteristics and the clinical information (Shang et al., 2015), we employ it as fitness function, which can be formulated as
where
Therefore, higher mutual information value denotes stronger association between the genomic characteristic combination and the clinical information.
2.2.3. Updating the dynamic scale-free network
A scale-free network, developed by Barabási and Albert, is a network whose degree distribution follows a power law. It is a loosely defined network topology that contains hub vertices with many connections, which grow in a way to maintain a constant ratio in the number of the connections versus all other nodes (Liu et al., 2014a; Gao et al., 2015).
In the basic PSO algorithm and most of the subsequent improved algorithms, it is generally considered that the status of each particle in the population is equal, thus neglecting the effect of the dynamic change of the population structure on the particle behavior. SIPSO and SF-PSO both use the scale-free network to simulate the population structure of particles. However, the scale-free networks used by them are generated in advance without considering the population characteristics of particles.
In general, updating the scale-free network incorporates two ingredients, including growth and preferential attachment. In the conventional scale-free network, randomly select new particles to join the network and choose neighbors for them with a specified probability that depends upon degrees of their candidate neighbors. In DSFPSO, we adopt a new strategy of growth and preferential attachment. In this study, we select new particles with higher fitness to join the network and choose excellent neighbors for them, which greatly improves the reliability of information exchange between particles.
In DSFPSO, the scale-free network building is synchronized with the solving iteration. At each iteration of network building, we sort the particles outside of the network in fitness descending order and select new particles with higher fitness value from these particles to join the network. Then we take particles that have already been in the network with the same sorting process and choose excellent neighbors for the new particles. In the dynamic process of scale-free network building, the particles' position and population structure will be dynamically adjusted with the joining of new particles to approach the best direction.
2.2.4. Updating the particle velocity
Since DSFPSO employs dynamic scale-free network as its particle population structure, the heterogeneity of particles and their neighbors has been greatly improved.
In the process of scale-free network building, particles have the difference of “in” and “out” of the network. For the particles in the network, take the first velocity updating strategy that considers the information of all neighbors and the inertial velocity to approach the best direction. For the particles out of the network, take the second updating strategy that considers the information of its own experience, the collective experience from group, and the inertial velocity. The velocity updating equations in the network building can be formulated as
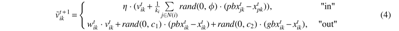
where
After the building of scale-free network, particles can be divided into two classes by the number of neighbors. According to a threshold, we decide different particles to choose the appropriate velocity updating strategy. For central particles, take the first velocity updating strategy that considers the information of all neighbors and the inertial velocity to make the optimum decision. For other particles, considering the information from its best neighbors, its own experience, and the inertial velocity, particle velocity updating becomes highly free and easy to search a wider space.
The velocity updating equations after the building of the network can be described as
where kc is a threshold that is used to decide the selecting of appropriate strategies to update the velocity for different particles.
Overall, different velocity updating strategies assigned to different particles not only consider the heterogeneity of particles but also effectively utilize the collective experience for scientific decision-making, which both ensure the dynamics of population structure and avoid local optimization.
Based on the mentioned velocity updating of particles, the position updating equations can be formulated as
An integer between 1 and M is randomly sampled if
2.2.5. Updating personal best position, neighbor best position, and group best position
In DSFPSO, personal best position of particle i will be updated by the position with the maximum mutual information. The specific equations can be formulated as
Similarly, the group best position updating equations can be written as
And the neighbor best position updating equations can be defined as
where
2.2.6. Finding final results
In the primary results of DSFPSO, a gene can appear at most four times when all four types of genomic features support the gene, so we resort scoring scheme to extract features (Liu et al., 2016). The scoring scheme by ranks gives priority to genes with top ranks or multiple ranks. The scoring function can be described as
where
In this study, the rank of each genomic feature is determined by the fitness value of particle personal best position in descending order. First, we calculate the unit score to weight the rank proportionally. Then, the score of top ranked genomic feature will be n times higher than the score of the bottom ranked genomic feature, which is shown in Equation (16). Finally, the score of a gene will be the sum of scores of genomic features from the gene, which is described in Equation (17). Therefore, the gene with a highly ranked genomic feature or with multiple genomic features will have a higher score overall and will be identified as the characteristic gene.
3. Results
3.1. TCGA data sets
We evaluate the performance of DSFPSO on two data sets: colorectal cancer (CRC) data set and cholangiocarcinoma (CHOL) data set.
CRC data set can be obtained from (https://tcga-data.nci.nih.gov/docs/publications/tcga). Data used in this article are the integrated data that has been preprocessed (Lee et al., 2013) (http://genomeportal.stanford.edu/tcga-crc/pages/datainformation). The CRC data of TCGA (Fig. 2.) used in this article from 197 samples contain 5188 genomic features of 1325 genes, including copy number variation, somatic mutations, methylation data, and gene expression data.

The CRC data of TCGA. CRC, colorectal cancer; TCGA, The Cancer Genome Atlas.
CHOL data set is a data set of 65,159 genomic features from 45 samples, including copy number variation, gene expression data, and methylation data. CHOL data,set is Level 3 processed, which can be obtained from (https://cancergenome.nih.gov).
We use a gene-centric approach for this study. For each gene, there are several different genomic features, such as copy number variation, mutations, DNA methylation, and gene expression. Since the primary multi-omics data are heterogeneous data, we carry discretization on these data sets that greatly improve the stability of the experiment. Then we integrate the data as a genome matrix. In the genome matrix, each row represents a sample, whereas each column represents a genetic characteristic. The whole genome matrix represents the search space of particles. In the clinical information matrix, the label of an individual is a binary phenotype being either 0 or 1.0 denotes control, whereas 1 denotes case.
3.2. Results on CRC data set
CRC is the second largest cause of cancer-related deaths in Western countries. Colorectal carcinogenesis is a multistep process involving apoptosis, differentiation, and survival mechanisms. To provide better treatment strategies, there is an urgent need to further understand the precise molecular mechanism of CRC and to identify new prognostic biomarkers and therapeutic targets for CRC (Li et al., 2015).
3.2.1. Gene ontology enrichment analysis
To verify the performances of DSFPSO, we carry out gene ontology (GO) enrichment analysis using ToppFun (https://toppgene.cchmc.org/enrichment.jsp) to find significant shared GO terms and compare the results with the state-of-the-art approaches.
On CRC data set, we input the top 500 genes identified by DSFPSO, PSO, SIPSO, LASSO, PMD, and SPCA into the ToppFun, respectively. The threshold value of the p value is set to 0.01 and other parameters are set as default.
Table 1 lists the top 10 closely related GO terms found by ToppFun. The lowest p value of every GO term has been marked in bold font. From this table, we can see that the term of “positive regulation of gene expression” (GO: 0010628) has the lowest p value (9.38E-19), so it is considered as the most probable enrichment item. It is reported that wild-type p53 mediates positive regulation of gene expression through a specific DNA sequence element and can regulate DNA methylation by cell-to-cell communication (Zambetti et al., 1992; Fuqua et al., 2001). We also find that most of GO terms identified by our method are positively regulated. Furthermore, we notice that in the term of “regulation of multicellular organismal development,” PSO outperforms DSFPSO, and in the term of “regulation of transcription by RNA polymerase II,” PMD outperforms DSFPSO. In general, DSFPSO shows better performance than SIPSO, PSO, LASSO, PMD, and SPCA in majority results.
Gene Ontology Enrichment Analysis on Colorectal Cancer Data Set Found by ToppFun
GO:0010628: positive regulation of gene expression; GO:0045595: regulation of cell differentiation; GO:2000026: regulation of multicellular organismal development; GO:0051254: positive regulation of RNA metabolic process; GO:1902680: positive regulation of RNA biosynthetic process; GO:1903508: positive regulation of nucleic acid-templated transcription; GO:0045893: positive regulation of transcription, DNA-templated; GO:0006357: regulation of transcription by RNA polymerase II; GO:0045935: positive regulation of nucleobase-containing compound metabolic process; GO:0051172: negative regulation of nitrogen compound metabolic process.
DSFPSO, particle swarm optimization-based algorithm with dynamic scale-free network; GO, gene ontology; LASSO, least absolute shrinkage and selection operator; PMD, penalized matrix decomposition; PSO, particle swarm optimization; SIPSO, selectively informed PSO; SPCA, sparse principal component analysis.
3.2.2. Gene function analysis
To describe the genes identified by DSFPSO, PSO, SIPSO, and LASSO and help discover what those genes may have in common, we study the overlap among the sets of top 100 characteristic genes identified by these methods on the CRC data set. The overlap among the sets of characteristic genes is presented in Figure 3. From this figure, we note that there are two genes shared by all the four methods and the characteristic genes identified by DSFPSO are to a large extent compared with the genes identified by PSO, SIPSO, and LASSO. Forty-four genes identified by DSFPSO are not shared with other methods.

The overlap among the sets of characteristic genes identified by DSFPSO, PSO, SIPSO, and LASSO on the CRC data set. LASSO, least absolute shrinkage and selection operator; PMD, penalized matrix decomposition; PSO, particle swarm optimization; SIPSO, selectively informed PSO.
To identify candidate genes with relevance to CRC, we review the biologic information of genes that DSFPSO can identify, but the others cannot (Liu et al., 2012; Va et al., 2014). Table 2 lists the biologic information of top 10 genes among the 44 genes by GeneCards and Entrez Gene Summary. Among these top 10 candidate genes, epithelial splicing regulatory protein 1 (ESRP1) is an epithelial cell-specific RNA binding protein, and splicing factor that identified as a tumor suppressor in the colon adenocarcinoma cell line (Leontieva and Ionov, 2009; Fagoonee et al., 2017) has found that ESRP1 plays a role in anchorage-independent growth of CRC cells. Moreover, ESRP1 promotes the ability of CRC cells to generate macrometastases. High ESRP1 expression may thus stimulate growth of cancer epithelial cells and promote CRC progression. Deloria et al. (2016) support the role of ESRP1 as tumor suppressor and strongly suggest that ESRPs are candidate markers for early detection, diagnosis, and prognosis of CRC. Nuclear receptor-interacting protein 2 (NRIP2) is a novel interactor of the Wnt pathway from enriched CRC colosphere cells (Pan et al., 2017). NRIP2 significantly promotes the self-renewal of colorectal cancer-initiating cells (CCICs) from both cell lines and primary CRC tissues. For the positive role in Wnt, NRIP2 may be a potential alternative target for inhibiting CCIC self-renewal (Wen et al., 2015, 2017). Potassium channel tetramerization domain containing 12 (KCTD12) is down regulated in the cells of CRC. Inhibition of endogenous KCTD12 and the overexpression of ectopic KCTD12 dramatically enhance and repress CRC cell self-renewal ability, respectively (Cheng et al., 2018). KCTD12 suppresses CRC cell stemness markers, such as CD44, CD133, and CD29, by inhibiting the ERK pathway. Li et al. (2016) reveal that KCTD12 is a novel regulator of CRC cell stemness and may serve as a novel prognostic marker and therapeutic target for patients with CRC (Luo et al., 2017).
The Biologic Information of Genes Only Identified by Particle Swarm Optimization-Based Algorithm with Dynamic Scale-Free Network
CRC, colorectal cancer.
3.2.3. Kyoto Encyclopedia of Genes and Genomes pathway analysis
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a database that systematically analyzes the function of gene to reveal the genetic and chemical blueprint of life (Kanehisa et al., 2008). In this study, we use the comprehensive set of functional annotation tools DAVID (https://David-d.ncifcrf.gov) on KEGG pathway to investigate the key pathways associated with CRC (Huang et al., 2009).
The top 10 KEGG pathways are given in Table 3. From the table, we can find that most enriched KEGG pathways are associated with cancer and we mark them bold in Table 3. Among them, “Pathways in cancer” and “Colorectal cancer” are obviously correlated with CRCs. “Transcriptional misregulation in cancer” is the major enriched pathway for upregulated transcripts in CRC (Dai et al., 2017). “HTLV-I infection” is associated with virus infection that may lead to cancer. Li et al. (2012) indicate that “PI3K-Akt signaling pathway” plays an important role in inflammation-induced colorectal carcinogenesis. “PI3K-Akt signaling pathway” links intimately with cellular metabolism and has great influence on cancer biological behavior (Josse et al., 2014).
Kyoto Encyclopedia of Genes and Genomes Pathway Analysis on Colorectal Cancer Data Set
3.3. Results on CHOL data set
CHOL is a devastating malignancy and is notoriously difficult to diagnose. The incidence of intrahepatic CHOL is increasing worldwide (Xu et al., 2010).
3.3.1. GO enrichment analysis
For GO enrichment analysis on CHOL data set, we put the top 1000 genes selected by DSFPSO, PSO, and SIPSO into ToppFun to test. And then we set p value to 0.01 and further analyze the performances of these methods.
Table 4 lists the top 10 closely related GO terms on CHOL data set found by ToppFun. The lowest p value of every GO term has been marked in bold. From this table, we can see that the term of “organic acid catabolic process” has the lowest p value (1.480E-16), so it is considered as the most probable enrichment item. Furthermore, we notice that in the term of small molecule catabolic process,” PSO outperforms DSFPSO. In general, DSFPSO shows better performance than SIPSO and PSO in majority results.
Gene Ontology Enrichment Analysis on Cholangiocarcinoma Data Set Found by ToppFun
GO:0016054: organic acid catabolic process; GO: 0046395 carboxylic acid catabolic process; GO:0019752: carboxylic acid metabolic process; GO:0044282: small molecule catabolic process; GO:0006082: organic acid metabolic process; GO:0043436: oxoacid metabolic process; GO:0051186: cofactor metabolic process; GO:0072329: monocarboxylic acid catabolic process; GO:0006732: coenzyme metabolic process; GO:0032787: monocarboxylic acid metabolic process.
3.3.2. KEGG pathway analysis
The top 10 KEGG pathways on CHOL data set are shown in Table 5. Several important metabolic pathways, such as “metabolic pathways,” “valine, leucine, and isoleucine degradation,” “glyoxylate and dicarboxylate metabolism,” and “propanoate metabolism,” are identified which are marked in bold in Table 5 (Leskinen et al., 2010; Chen et al., 2015).
Kyoto Encyclopedia of Genes and Genomes Pathway Analysis on Cholangiocarcinoma Data Set
4. Conclusions
Considering traditional PSO algorithms usually takes equal treatment of all particles and ignores the advantages related to the heterogeneity of population structure, an improved PSO named DSFPSO is proposed for detecting multi-omics features. In DSFPSO, with fitness value of particles as a standard for link growth and preferential attachment, the scale-free network building is synchronized with the solving iteration. Moreover, this work is a first study of multi-omics feature detection with an improved PSO algorithm. We carry out the experiments on two data sets provided by TCGA and find the final results through scoring strategies. Experimental results show that DSFPSO can converge to global optimization quickly and achieve more comprehensible and interpretable results, which will provide valid references for early diagnosis, effective treatment, and prognostic guidance of cancers.
Maintaining the links between particles may need to consume considerable time. So in future, we will work on exploring correlations among differentially expressed genes and try to pruning the extra links that do not have much effect on the information change between particles. And we will also concentrate on improving the algorithm, exploring correlations among differentially expressed genes, and looking for the key regulatory factors for the tumor process as well as the potential therapeutic targets for patients.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant nos. 61502272, 61572284, and 61873331), Project of Shandong Province Higher Educational Science and Technology Program (J18KA373), and the Science and Technology Planning Project of Qufu Normal University (xkj201524).
Author Disclosure Statement
The authors declare that no competing financial interests exist.