
Abstract
Abstract
1. Introduction
One of the most fundamental tasks in biology is deciphering the history of life on Earth. Darwin was the first to speculate that the history of life is best described using a tree structure. The leaves of this tree represent existing species and the branches represent evolutionary relationships. It is of no wonder that Darwin's only drawing in his On the Origin of Species (Darwin, 1859) is a sketch of a tree structure. A direct implication of this theory is that all life forms evolved through a series of mutations, occurring in an ancestor–descendant basis since the beginning of life.
This is the predominant theory of evolution, and hence, its appeal to exact sciences. In 2006, this theory was put by Valiant (2006, 2009) under a rigorous computational framework attempting to explore the learning power of this mechanism. A genome is viewed as a function, reacting to a set of signals from the outer world. Over time, this function improves toward a hidden ideal function, through generating mutations and through natural selection. Valiant named this framework evolvability. The goal of computational learning theory is to separate concept classes that can be efficiently learned under a certain framework, from those that cannot. Then, the formal question asked by Valiant is, what is the complexity of functions that can be efficiently learned. Examples of concept classes of interest include Conjunctions, Parities, and so on. Thus, the question of evolution can be asked in the language of computational learning theory: For what classes of functions, can one expect to find an evolutionary mechanism that gets arbitrarily close to the ideal, within feasible computational resources?
All previous works, except for the recombination model of Kanade, assumed a vertical mode of evolution where variation is introduced by applying random mutations to the parent's inherited genome. However, the emergence of high-throughput sequencing revealed strong signals that stand in conflict to the Darwinian theory. It appears that some life forms on Earth do not adhere to the principals of vertical evolution. Specifically, prokaryotic evolution is characterized by extensive gene mobility between species that is crucial for their survival (Koonin and Galperin, 2002; Gogarten and Townsend, 2005). The principal mechanism accounting for gene mobility is horizontal gene transfer (HGT) (Doolittle, 1999; Ochman et al., 2000; Koonin et al., 2001; Nakamura et al., 2004; Jin et al., 2007) in which a group of genes can be transferred from a donor organism to recipient. Recent studies have shown that HGT stands out as the dominant factor in several important phenomena in bacterial evolution: adaptation to niches, development of antibiotic resistance, and pathogenicity. Even though HGT is an important factor in prokaryotic evolution, our knowledge on HGT is very limited, partly due to the lack of use of analytic tools and models in the field (Doolittle and Bapteste, 2007).
In this work, we extend the Evolvability theory to account for this less studied, yet very important, mode of evolution. In particular, we use the notion of population-based models where a population is evolving simultaneously and information is shared between members of this population. The model is inspired by the basic evolvability model and is extending it. The new model accommodates mechanism such as HGT where genetic information in the form of DNA is passed between individuals. While in a recent separate work (Snir and Yohay, 2018), we focused on the theoretical aspects of the newly defined model and showed that it provides an asymptotic acceleration in learning, here the emphasis is on more biological practical points. We first give the formal necessary definitions as required by the new extension. Subsequently, we use the conjunction function to model the process of acquiring a character or a property by an organism. Focusing on this important function class, we show experimental results regarding the new and existing models. Finally, we analyze real data in the form of developing virulence in Escherichia coli. We demonstrate that this function can be depicted by a monotone conjunction and compare between the models on this data. Our results, in the form of actual concrete times, give convincing evidence to the superiority of the HGT process over more conservative mechanisms, explaining the vastness of the gradually discovered world of mobile elements—the mobilome (Siefert, 2009).
2. Preliminaries
The new extension, which was first defined in our recent work (Snir and Yohay, 2018), relies on the existing theory of evolvability. We now give necessary definitions required for the extension. For the sake of compatibility with previous works, we tried to use original definitions from existing works in places where this was possible. In such cases, we give explicit reference to the appropriate work inside the definition.
2.1. The evolvability model
A description of the evolvability framework is necessary for our extension and is given hereby. For the full definition of the evolvability model, the reader is referred to the work of Valiant (2009).
We start with a notational comment. Throughout the article, we abuse notation
We assume a finite set of conditions organisms have to respond to. A condition may represent a certain disease, availability of food, or water. All the possible combinations of conditions are given by the set
The functions discussed in this work are Boolean functions with domain X and possible outputs of −1 and +1. A concept class C over X is a set of Boolean functions. Suppose that
The hypothesis shall be viewed as a representation of a function since it should be represented concretely in the organism. In accordance with the function class, a representation class R is a set of representations, such that every
The goal of the evolvability learning algorithm is to evolve some representation into a hypothesis that gets arbitrary close to the ideal function, using evolutionary processes. The learning occurs throughout discrete generations.
2.2. Evolvability with HGT
We now define a model that captures the central mechanisms in the process of HGT.
The neighborhood function is the algorithm by which representations mutate across generations. We define a neighborhood function such that a representation can receive information (genes) from many different representations:
Thus, r is mutated according to the set
We now define the performance. The performance is used to give a quantitative measurement of the how well a representation approximates the ideal function.
The evolutionary algorithm will have access to an oracle that given a representation returns its estimated performance. We define the estimated performance according to Valiant's basic evolvability definition, with a slight modification: Instead of observing the sample size s, we observe the estimation error (noise)
We now define the tolerance function. The tolerance function separates the mutations that performed good from the other mutations in relation to the current representation's performance.
We define a probability function
A selection rule Sel selects a (possibly random) mutations of the neighborhood function based on its estimated performance. In this work, we use the selection rule used by Valiant, which we denote by SelNB.
Let If Otherwise, if Otherwise, output
Thus, SelNB chooses a beneficial mutation if one exists or otherwise chooses a neutral representation.
The notion of a population was suggested by Kanade (2011) to allow the process of recombination between individuals. Indeed, in an environment where information is shared between organisms, it is more natural to look at a set of representations across generations. Thus, we assume the existence of a finite population with polynomial size.
An HGT mutator (or evolutionary step) takes a population Pi to population
Let
While
Select randomly Consider the mutations Activate the selection rule function If
Finally, we define the notion of evolvability with HGT in g generations.
In our recent work (Snir and Yohay, 2018), we have showed the equivalence between evolvability and evolvability with HGT, by proving the following straightforward theorem:
The equivalence shows that the models can learn the same range of concept classes efficiently (i.e., within polynomially bounded resources). Nevertheless, different models can learn the same problem in different speed, that is, different number of generations. The main result of our previous work (Snir and Yohay, 2018) manifests that the HGT extension allows an acceleration in terms of the number of generations. This is performed using a general reduction from the parallel CSQ model (Correlational Statistical Query, a learning model that performs queries like PAC) that we define below.
2.3. Parallel CSQ models
We define a model for parallel correlational statistical-query learning with a
In our recent work, we have proved a theorem that shows the power of the HGT model using a general reduction from parallel CSQ model.
This theorem shows that from a theoretical perspective, the HGT model allows to learn in less generations than possible using the basic model. Next, we describe experiments we devised that compare between the different learning models. The experiments show the same trend.
3. Results
In this section, we show several implications of the theoretical results described in previous sections. We model a biological trait that depends on multiple parameters as a conjunction function and acquisition of that trait as learning of the function. Due to the importance of this class, we derived analytical results in a recent article (Snir and Yohay, 2018) and now strengthen these results with experiments. We run simulations in which the conjunctions concept class is learned and show that these results affirm our analytical results. We end this section by applying these models to real biological data regarding the development of pathogenicity in microbes.
3.1. The conjunction function and concept class
Let the sample space consist of n Boolean variables (literals),
The concept class of monotone conjunctions is the set of conjunction classes such that the classes do not contain a negated literal.
Conjunction is biologically relevant as many biological processes or characters can be seen as a result of the simultaneous existence of a set of genes and absence of another set (see, e.g., microbial pathogenicity in our real data part Section 3.3). The same also holds for an expression of a certain protein that is conditioned on the existence and absence of some other proteins (Alon, 2006).
The values
3.2. Simulation study
We conducted simulation study to illustrate the difference between the various models under realistic size problems. The processes (models) examined are as follows: the mutation process (i.e., evolvability), recombination, and HGT. As each such process uses another technique to adapt to the environment, we ask, how fast, in terms of generations, a given function is learned, under each such evolutionary process.
Roughly speaking, we can divide the models into two very distinct groups: individuum-based model, where only a single representation is examined, and population-based models, where a population of representations are generated, each generation and relations between them are enabled. Even though recombination and HGT are population-based, recombination allows merging only between two individuals, whereas HGT allows information sharing between the whole population. We will see that this variance makes a large difference in the results.
In reality, HGT is mandated by HGT rate that determines probability of HGT events. Therefore, the first experiment measures the effect of HGT rate on the speed of evolution. We model the HGT as a Poisson process (Grimmett and Stirzaker, 2001) operating on a genome through time (Galtier, 2007; Roch and Snir, 2012). This allows easy conversion from rate to event probability. We executed the HGT algorithm with HGT rate varies from
In the second experiment, we examined the interplay between the two processes occurring in the population simultaneously. Underneath the population processes—recombination and HGT—an underlying mutation process operates individually on every element. We therefore set to test that interplay under the two population models. Specifically, we varied the underlying mutation factor while maintaining all other parameters constant, including the population parameters.
The third experiment deals with the role of the size of the population in learning. We start with some population and increase it up to a size of five times that starting population.
3.2.1. Learning and models description
We have conducted the experiments with three models: the basic evolvability model described by Valiant, evolvability with recombination as described by Kanade, and the model we described in Section 2.1, evolvability with HGT. We start by describing the common setup and goal of the models. Our overall goal is to draw a distinction between the three learning models. As the learning processes are computationally very heavy, we selected fairly small parameter values; however, the general trend is still reflected.
The parameters in the experiments are thus: The number of Boolean variables was set to 40 (
A run of the experiment starts by choosing a random ideal function f and a representation r (or a population of such) and trying to learn f throughout generations. The next generation is obtained by applying the mutator (or recombinator) to the current generation.
The learning stops when at least one representation r in the current population satisfies
3.2.2. Experimental results
We now describe the experiments performed. In any experiment, the number of runs was set to 10 for any value of the independent parameter (x) and the average result (generations) is plotted.
Our first experiment focused on the effect of HGT on the speed of learning. Obviously, this parameter is effective only to the HGT model. In the population-based models, mutation factor was set to 0.1, so it does not interfere with the overlying processes recombination and HGT (in the basic evolvability model, mutation is the only process so we set it to 1). The results of the experiment are shown in Figure 1. With an HGT rate of 0.2 or higher, the HGT model outraces the other models. Actually, under HGT rate of 1, the model learns in almost third of the generations that took the recombination model. Alternatively, under HGT rate of 0, learning is confined only to natural mutation with a factor of 0.1, which explains why it is very slow, even slower than the basic evolvability model. We can therefore infer that HGT rate plays a major role in the learning process of the HGT model.

In the next experiment, we examined the role of the underlying mutation factor under the three models. For HGT, we considered two rates: 0.2 and 1. In the experiment, mutation factor varied from 0 to 1. The number of Boolean variables of the functions is set to 10 and
Our final experiment examined the effect of population size on speed of learning under the two population models—recombination and HGT. We therefore varied population size from 100 up to 1000. The results (Fig. 2) show interestingly a constant decrease in learning time (generations) under both population models; however, HGT decreases faster than recombination.

Experimental results of the effect of the size of the population on the number of generations.
3.3. Real data analysis
In this section, we apply the same models from the previous section on real data. We chose to focus on the pathogenicity of the E. coli bacteria. The virulence of an organism is the degree of pathology caused by that organism. To use the evolvability framework, we need a quantitative trait of pathogenicity, which is why we focus on virulence in this section. A virulence gene is a gene whose existence in the genome of an organism affects its virulence. A genome is considered pathogenic if it has an appropriate virulence gene combination (Chapman et al., 2006).
We do not, however, claim that these models represent the processes exactly as they occur in nature. The use of real data in the rigorous framework of evolvability and the comparison between the models grants a realistic aspect to this framework and, in particular, to evolvability with HGT, and hence its importance.
We now show an example of how to deduce the Boolean variables from the genes: The pathogenic strain Enterotoxigenic E. coli was identified by Do et al. (2005) by carrying either the gene combination fedA, estII or faeG, estI, estII, eltA. First, we enumerate the genes
3.3.1. Simulation of real data results
We compare between the power of two biological processes: the process of HGT and recombination.
Fifty-eight virulence genes were observed in the work of Chapman et al. (2006). The number of Boolean variables was set to 100, where 58 of them were randomly chosen to represent the virulence genes and their values were set to 1. The other variables were chosen randomly. A value of 1 in one of the other variables represents a virulence gene that has yet been discovered.
The size of the population is chosen to be 75, corresponding to the 75 E. coli isolates that were taken in the work of Chapman et al. (2006). The approximation parameter
An estimation of the HGT rate for E. coli showed that 17.6% of the genes have been transferred since its divergence from Salmonella lineage 100 million years ago (Lawrence and Ochman, 1998). We consider a single generation in our models as a million year and set the HGT and recombination rate to be 0.176% (A recombination rate of x signifies that if recombination does not occur, one of the two representations is chosen randomly).
Over a total of 20 runs, the recombination model learned in 256 generations average, and the HGT model learned in 35 generations average.
In conclusion, if evolution would have occurred according to our parameters, a highly virulent organism would have emerged in 35 million years using HGT process. If instead the recombination process would have taken place, the pathogenic organism would have emerged in 256 million years.
3.3.2. Comparison with Escherichia lineage
In this section, we try to measure how the result from the previous section compares with real data. The evolvability with HGT model yielded a 35 million years for a highly virulent pathogen to emerge. We have put this result alongside the divergence times in the evolutionary lineage of Escherichia (Walk et al., 2009). The evolutionary lineage is displayed in Figure 3. Interestingly, the result is in the same order of magnitude as the splits in the evolutionary lineage.
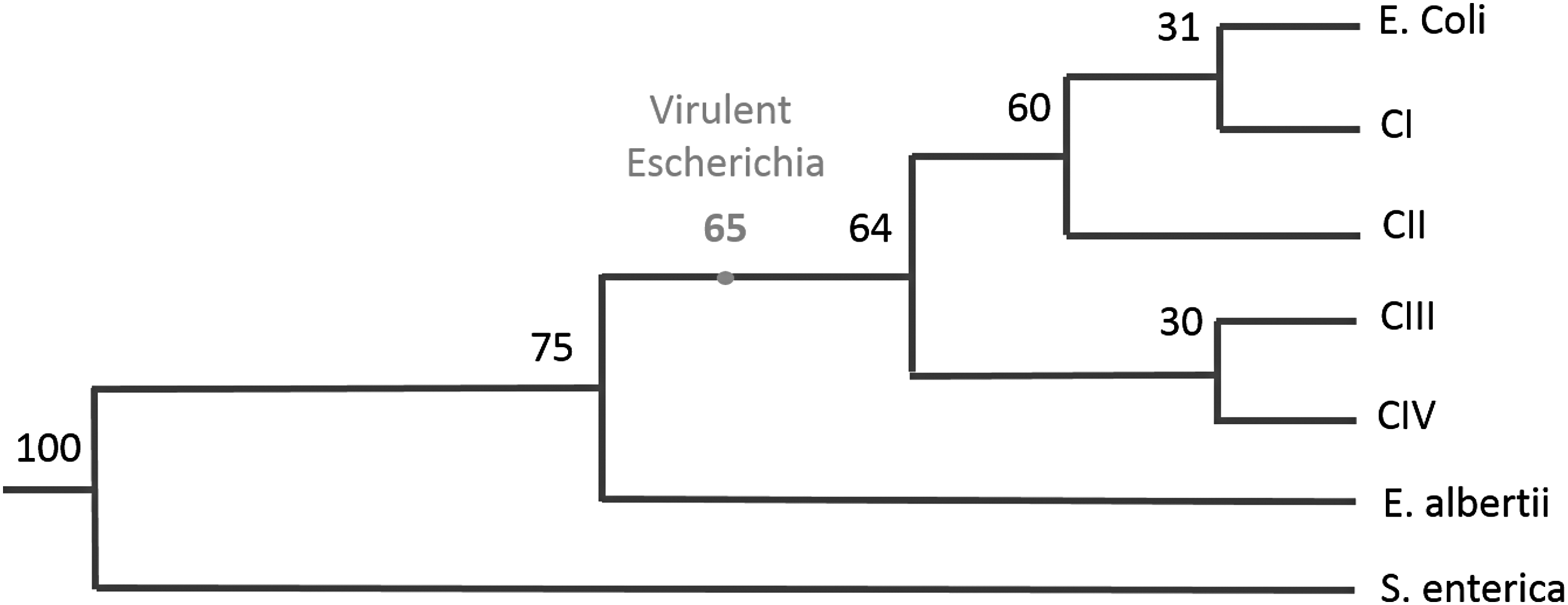
Evolutionary lineage of the Escherichia genus. The nodes indicate how long ago the divergence occurred in million years. The dot indicates the result that evolvability with HGT yielded: The emergence of a highly pathogenic organism, starting from the divergence from Salmonella.
4. Conclusions
In his seminal work from 2006 (Valiant, 2009), Leslie Valiant extended his celebrated PAC (Probably Approximately Correct) model (Valiant, 1984) to the biological world and denoted it evolvability. Evolvability quantifies the process of evolution in terms of computational learning power. Subsequently, Kanade (2011) extended evolvability to higher level organisms' reproduction by combining the mechanism of recombination. Nevertheless, to the best of our knowledge, no such extension to evolvability was suggested for prokaryotes. Prokaryotic evolution largely proceeds by exchanging DNA between unrelated organisms, a mechanism denoted HGT. It is mainly due to HGT that microbes adapt to new ecological conditions, develop resistance to antibiotics, and so forth. In this work, we define a conceptual model encompassing this phenomenon. The model is conceptual/mathematical and therefore is not limited to a specific biological phenomenon. Valiant's evolvability is a special case in this new model. The evolutionary advantage of the new model is by allowing (genetic) information sharing between individuals in an evolving population, similar to the recombination model.
While in a recent work, we have focused on the theoretical aspects of the newly suggested model, here the emphasis is on its biological application. Specifically, we focus at apparently a biologically relevant function—conjunction, modeling existence of an entity contingent on the presence of several other entities. We show that for conjunction, the new model achieves asymptotic acceleration in learning time over evolvability and recombination. We corroborate these findings in simulation where we use a randomized learning algorithm for conjunction under evolvability. We conclude with learning a real data function of virulence in the E. coli bacterium.
We believe that the importance of this model stems from the lag of application of rigorous computational learning tools to model evolution in this life domain. The comparison between several modes of real-life evolutionary mechanisms under a common ground, from a rigorous computational learning perspective, provides another explanation to the versatility of this increasingly discovered world.
There are several future directions to take from this work that we consider of interest. Conjunction may be relevant for the case discussed here, but other biological mechanisms may require other concept classes. This may give insight into these mechanisms. We think that evolvability is a powerful yet flexible tool and can be used extensively to analyze more real biological data, using the scheme described in this work. Finally, modeling more evolutionary phenomena with this framework is interesting both from the aspect of computer science and from the aspect of biology.
Footnotes
Acknowledgment
The authors wish to acknowledge the Israel Science Foundation (ISF) for its kind support in performing this research.
Author Disclosure Statement
The authors declare that no competing financial interests exist.