
Abstract
Inframe insertion and deletion mutations (indels) are commonly observed in cancer samples accounting for over 1% of all reported mutations. Few somatic inframe indels have been clinically documented as pathogenic and at present there are few tools to predict which indels drive cancer development. However, indels are a common feature of hereditary disease and several tools have been developed to predict the impact of inframe indels on protein function. In this study, we test whether six of the popular prediction tools can be adapted to test for cancer driver mutations and then develop a new algorithm (IndelRF) that discriminates between recurrent indels in known cancer genes and indels not associated with disease. IndelRF was developed to try and identify somatic, driver, and inframe indel mutations. Using a random forest classifier with 11 features, IndelRF achieved accuracies of 0.995 and 0.968 for insertion and deletion mutations, respectively. Finally, we use IndelRF to classify the inframe indel cancer mutations in the MOKCa database.
1. Introduction
Most cancers are formed as a result of genetic mutations in DNA sequences in critical genes that confer a selective advantage to tumor cells (Futreal et al., 2004). These coding mutations can be caused by error in DNA replication and repair, and environmental factors that alter the genetic structure of somatic cells. Understanding the impact of these mutations is vital for providing a platform to understand cancer initiation, progression, and therapeutic strategies (Ferrer-Costa et al., 2004; Hindorff et al., 2009).
Commonly observed somatic variations in cancer include single-nucleotide variants (SNVs) and small insertions and deletions (indels). Indels are the second most common type of mutations after SNVs with over two times as many deletions as insertions occurring in most cancers (Stenson et al., 2009). Indels can affect protein function and contribute to cancer development (Akagi et al., 2010).
Two types of indels are found in protein coding regions, frameshift and inframe mutations. Indels that cause frameshifts have a length not divisible by three, they change the reading frame of the DNA. They generally result in a change to the amino acid sequence, followed by a premature stop codon and a truncated transcript. Indels that have a length divisible by three are called inframe indels and cause insertions and deletions of small runs of amino acids (Mullaney et al., 2010).
Cancer mutations, including indels, are considered driver mutations if they give the cells a selective growth advantage and contribute to the initiation or progression of the disease. Passenger mutations do not contribute to the disease progression per se, but occur due to the inherent genetic instability of the tumor (Greenman et al., 2007). Driver mutations that contribute in tumorigenesis are normally found in genes described as oncogenes or tumor suppressor genes depending on their role in cancer development (Futreal et al., 2004).
Although the majority of computational tools developed for assessing genetic mutations have focused on missense mutations, more recently, there have been several efforts to predict the impact of inframe indel mutations on protein function or structure using a variety of strategies. Commonly used algorithms that predict the pathogenicity of impact of indels include PROVEAN (Choi and Chan, 2015), SIFT (Hu and Ng, 2013), VEST-Indel (Douville et al., 2016), CADD (Kircher et al., 2014), DDIG-In (Zhao et al., 2013), PaPI (Limongelli et al., 2015), and PinPor (Zhang et al., 2014). Most of these methods classify each mutation according to two state categories, neutral or pathogenic, using a variety of machine learning techniques, including a J48 Decision Tree (SIFT-indel), Random Forest and Logistic Regression (PaPi), and Bayesian networks (PinPor), with reported area under the curve of receiver operating characteristic (AUC ROC) accuracies varying from 0.75 to 0.9 on a variety of data sets.
The pathogenic mutations in these algorithms are generally sourced from The Human Gene Mutation Database (Stenson et al., 2009), a catalog of gene lesions responsible for human inherited disease (e.g., SIFT-indel, VEST-indel, DDID-In, PaPI, PinPor), or from UniProt (PROVEAN). Neutral mutations are generally derived from the 1000 Genomes Project (Genomes Project et al., 2010), the Exome Sequencing Project (ESP) (Tennessen et al., 2012), or by identifying tolerated mutations from the sequence alignment of human sequences with other mammalian species (e.g., SIFT-indel). CADD uses a slightly alternative approach that discriminates fixed or nearly fixed derived alleles in human from a set of simulated mutations. This method was developed to predict deleterious mutations rather than the functional effect on protein or variant pathogenicity using a support vector machine (SVM) classifier (Kircher et al., 2014).
In this study, rather than studying genetic mutations from model organisms and inherited disease genes, we wanted to develop a method for determining driver indel mutations specifically for somatic mutations in cancer. However, few insertion and deletion mutations have been clinically documented as pathogenic in cancer. For instance in the ClinVar database (Landrum et al., 2014), only 20 inframe insertions and 108 inframe deletions are described as pathogenic and there were even fewer reported somatic driver mutations (8 and 26, respectively).
Recurrence is often used to imply clinical driver status to cancer mutations (Landrum et al., 2014). So, to identify a set of somatic indel mutations that were likely to contribute to the development of cancer, we decided to use recurrence. We identified a set of recurrent somatic indels found in exome sequencing of documented cancer genes. We investigated the ability of present prediction algorithms to distinguish between these recurrent mutations and neutral indel mutations found to have little or no effect on protein function. We then defined an “optimal” training set of cancer mutations that could be used in algorithms that predict whether an indel is contributing to the development of cancer.
An automated classifier was developed to distinguish between deleterious and neutral mutations using 11 features to describe each mutation. We selected a random forest classifier that achieved the best result to classify pathogenic and neutral mutations for insertions and deletions, respectively. We validated our approach by testing our algorithm using indels clinically identified as disease causing deposited in the ClinVar database. Finally, we ran our algorithm (IndelRF) classifier to classify the predicted inframe indels in the MOKCa database into pathogenic or neutral mutations.
2. Methods
2.1. Data
To identify recurrent mutations, inframe insertions and deletions (indels) were extracted from the COSMIC database v82, using annotations from the Ensembl human genome build hg38 (Bamford et al., 2004). Mutations were also extracted for the hg37 build of the Ensembl database for use with the PaPI, DDIG-in, and PinPor algorithms.
Clinically determined cancer mutations were downloaded from the ClinVar database (Landrum et al., 2014) with indels that were labeled as pathogenic or probably considered pathogenic.
For the neutral set of mutation, we identified a set of indels derived from the 1000 Genomes Project and the ESP that are commonly observed in the human population (Hu and Ng, 2013). To make sure that our trained data sets were balanced, no more than 10% of the mutations within a class were taken from a single protein or a domain type.
2.2. Identification of hotspot indel mutations
To identify indels that were likely to be pathogenic, we identified hotspot mutations. For each protein in the human exome, we computed the total number of mutations it contained and the frequency of mutation at each position. A binomial test was used to identify which positions had a significant number of mutations (Supplementary Material). Insertion and deletion were tested independently and only positions where mutations occurred at least twice were analyzed.
2.3. Comparison of prediction algorithms
We assessed six different algorithms that have been developed to predict the impact of inframe indel mutations on the protein function and structure. These algorithms were CADD (Kircher et al., 2014), DDIG-In (Zhao et al., 2013), PaPI (Limongelli et al., 2015), PinPor (Zhang et al., 2014), SIFT-indel (Hu and Ng, 2013), and VEST (Douville et al., 2016).
2.4. Feature selection
We derived features from four existing prediction algorithms: VEST, PinPor, CADD, and Pseudo Amino Acid Variant Predictor (PaPI). In total, we calculated 11 features for each mutation (Supplementary Table S1). These features describe the evolutionary conservation of the sequence where the insertion or deletion occurs, in a variety of ways, or the pathogenicity of the mutation.
2.5. Feature importance
Mean decrease accuracy was measured to identify the variable importance using the randomForest package (Archer and Kimes, 2008). The values of each of the variables in turn are randomly permuted for the out-of-bag observations, and then, the modified data are passed down the tree to get new predictions. The importance of the variable is the difference in misclassification rate for the modified and original data, divided by the standard error (Supplementary Material).
2.6. Machine learning
All data sets were balanced to remove protein and domain biases in the data set. No more than 15% of mutations were allowed from a single protein or a domain family. A random forest classifier was trained to classify pathogenic and neutral indel mutations using R version 3.2.3. Binary classifications were calculated for inframe insertion and inframe deletion, independently. It was run with 10-fold cross-validation, and the parameters optimized for each model.
We also trained an SVM classifier using 10-fold cross-validation to optimize the hyperparameter C, used to trade-off between variable minimization and margin maximization, and chose the kernel type that best fits our data.
The classifier with the best accuracy at discriminating between pathogenic and neutral mutations for both insertions and deletions was a random forest machine classifier that we have named IndelRF.
2.7. Validation of algorithms
We validated the performance of IndelRF and compared it with existing algorithms using test sets from the ClinVar database (Landrum et al., 2016). Predictions were generated using standard settings and the public web servers. Sensitivity (TP/TP+FN), specificity (TN/TN+FP), and accuracy (TP+TN/TP+TN+FP+FN) were measured to compare the performance of methods. We also calculated AUC of ROC for insertions and deletions separately.
2.8. Prediction of functional consequences of indel mutations in the MOKCa database
A total of 5437 inframe indel mutations were downloaded from MOKCa database v21 (Richardson et al., 2009). A total of 1167 of them were insertions and 4270 were deletion mutations. IndelRF was used to predict whether the mutations were pathogenic and likely to be cancer drivers. We also identified the pathogenic mutations found in oncogenes and tumor suppressors as described by the Cancer Gene Census (Futreal et al., 2004).
3. Results and Discussion
3.1. Identification of recurrent indels
A total of 4435 inframe insertion mutations and 14,456 inframe deletion mutations were reported in the COSMIC database. This led to 909 recurrently mutated positions having inframe insertions and 2587 inframe deletions. As more than one indel could be reported at each amino acid position in total, there were 1856 inframe insertions and 2766 inframe mutations that we used to compare the performances of the six published algorithms.
3.2. Comparison of prediction algorithms
3.2.1. Ease of use
The number of results successfully calculated by the prediction algorithms, for each of the insertion and deletion mutations, is shown in Supplementary Tables S2 and S3 and Supplementary Figure S1. Clearly, the algorithms did not work on all COSMIC annotations of the mutations. Often the reason was incomplete nomenclature. For instance, missing bases in the input sequences for deletions caused some algorithms to falter. The entries CTNNB1 c.14_241del228 and FOXP1 c.1553_1564del12 did not give results, as the sequence of the deleted DNA was absent from the entry.
There may have also been discrepancies in genomic location of the mutation that was required for the programs due to differences in versions of the genome build used to define the mutation and that the prediction algorithm used.
3.2.2. Are recurrent mutations pathogenic?
In total, pathogenicity values could be calculated for 898 inframe insertion and 962 inframe deletion predictions for all six programs available (Supplementary Fig. S1). The algorithms predicted between 27% and 62% insertion mutations and between 33% and 73% deletion mutations as pathogenic. In total, 74 inframe insertion and 109 inframe deletion mutations were predicted as pathogenic by all six algorithms (Fig. 1). DDIG-in predicted the least number of indels to be pathogenic, whereas PaPI identified the most number of indels to be pathogenic.
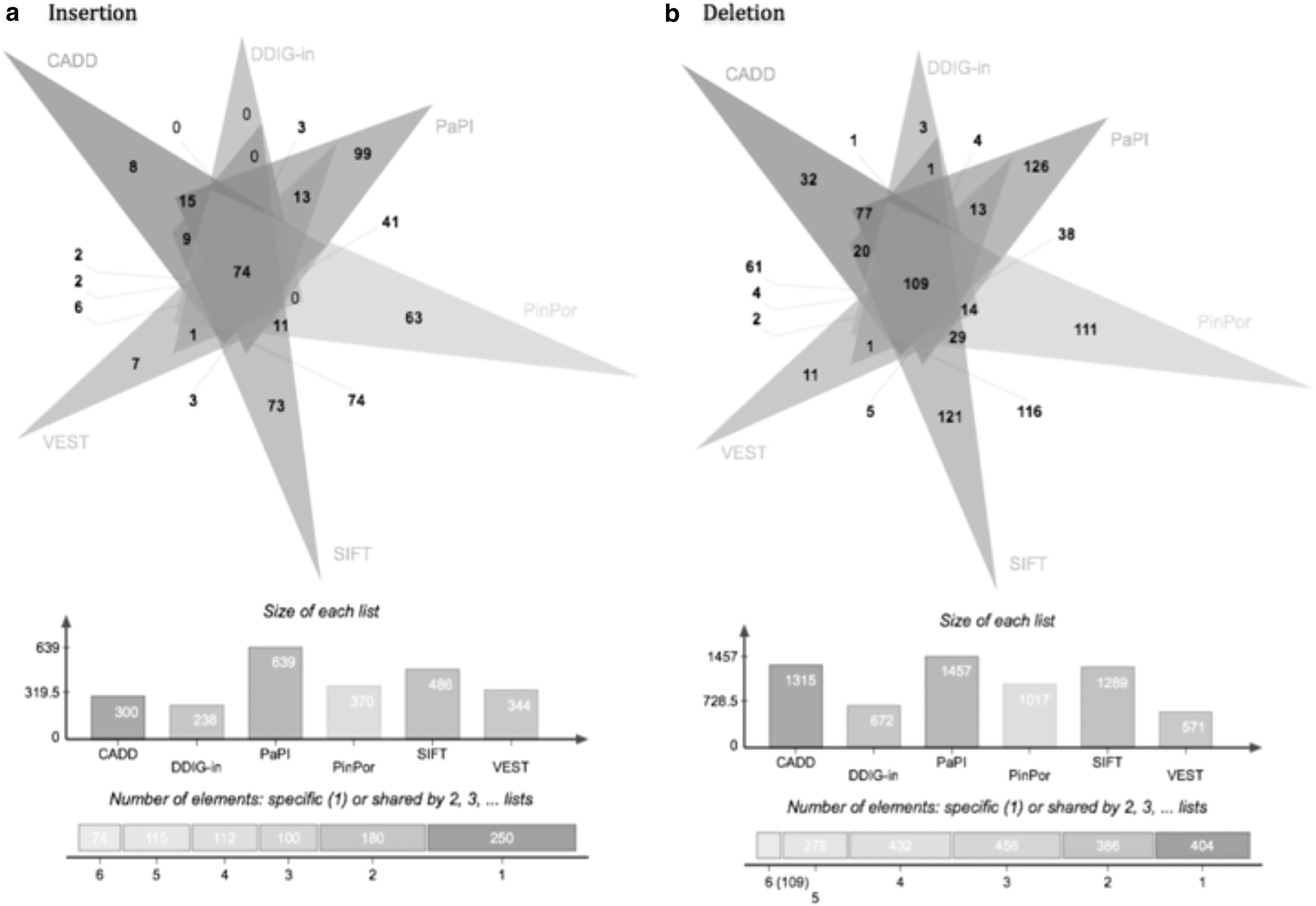
Common pathogenic mutations between six algorithms in inframe indels.
3.2.3. Definition of optimal somatic cancer pathogenic indel data sets
To compare the variation between the algorithms, we selected 98 recurrent insertion mutations and 155 recurrent deletion mutations, predicted to be pathogenic by at least four of the six programs, as our putative pathogenic driver indel data sets. This reduction in the number of mutations was to remove protein and domain biases in the data set so that no more than 15% of mutations within a data set were allowed from a single protein or a domain family.
When using the algorithms to distinguish between our somatic driver pathogenic indels and a neutral set of mutations, most of the algorithms performed well with accuracy scores ranging from 0.753 to 0.988, and similarly to their published performances on indels linked to hereditary disease (Table 1). The DDIG-in algorithm performed the best on these examples, discriminating well between the recurrent somatic cancer mutations and the neutral mutations for both inframe indels (Hu and Ng, 2013). The only exception was PinPor that had accuracy scores of 0.534 for insertions and 0.553 for deletions. PinPor differs from the other prediction algorithms as it predicts the pathogenicity of indels by assessing the impact of mutations on posttranscriptional regulation rather than impact on the protein structure.
Comparing the Performance of Inframe Insertion and Deletion with Previously Published Results
Acc, accuracy, AUC, area under ROC curve; NA, not applicable; Sen., Sensitivity; Spe., specificity.
3.3. Development of a cancer-specific indel classifier
Evaluation of our data sets by existing algorithms suggests that the recurrent somatic cancer mutations are pathogenic and therefore may be drivers in cancer. We then used these cancer-specific data sets to train machine algorithms to enable us to detect other driver indel mutations. Two different models, random forest and SVM classifiers, were trialed and compared. Binary classifications were calculated for pathogenic/neutral classes in inframe insertion and deletion, independently. We used a random forest classifier using 10-fold cross-validation to optimize classifier hyperparameters and assess performance for each class. The random forest classifier has two parameters, depth and number of trees, that affect the accuracy of a classifier (Bosch et al., 2007). The results show how the changing of both the number of trees and the depth of these trees affects the accuracy (Supplementary Tables S6 and S7), however, the classification accuracy is generally high. The highest accuracy is 0.995 when the depth is 100 and the number of trees is 100 in insertion mutations, and the highest accuracy is 0.968 when the depth is 10 and the number if trees is 1000 for deletion mutations.
We also trialed an SVM classifier, however, all our random forest classifiers performed better than our SVM classifier for insertion and deletion mutations. We found the highest accuracy of 0.983 and 0.962 with a radial basis function (RBF) kernel for insertion and deletion, respectively. The RBF kernel is the simplest kernel that can be used and generalizes good results (Suykens and Vandewalle 1999; Keerthi and Lin, 2003). The SVM classifier yielded the best result using the RBF kernel. The results for the SVM hyperparameter optimzation show that different values of hyperparameters in insertion and deletion mutations do not significantly change accuracy scores except when the polynomial kernel is used, which caused the classifier to have a lower accuracy of 0.658 and 0.654, respectively (Supplementary Tables S8 and S9).
3.3.1. Feature importance
Having successfully designed an algorithm that could reliably distinguish between recurrent somatic cancer mutations and neutral insertion/deletion mutations, we decided to identify the important features. Mean decrease accuracy is one of the popular feature selection methods that directly measure the effect of each feature on the accuracy of random forest. It permutes the values of one feature while others are left unchanged and measures how much the permutation reduces the accuracy (Cutler et al., 2007).
Figure 2 shows that VEST p-value, priPhCons, Phylop, and Gerp++ were the four best performing features for insertion and deletion. The VEST p-value score, from VEST prediction algorithm, is a measure of how likely a gene mutation is to be pathogenic. Primate PhastCons conservation score (priPhCons) was one of the top five features from CADD. Phylop and Gerp++ scores, from PaPI algorithm, are two of the evolutionary conservation scores that apply different and complementary methods to weight nucleotide conservation among different species (Garber et al., 2009).

The importance features across insertions and deletions. The features are ranked according to insertion mutations with the corresponding key at the side.
Moreover, the distance of indel mutation to the exon's 3′ end was one of the most important features for insertions. Similarly, when comparing pathogenic versus neutral mutation for deletions, one of the top five features was the distance of indel to exon's 5′ end.
3.3.2. Evaluation test set
We applied our algorithms to the pathogenic insertion and deletion mutations identified in the ClinVar databases as an independent evaluation set. For somatic insertion indels, 18 pathogenic mutations and 7 somatic pathogenic mutations were evaluated using (IndelRF) with accuracies of 0.833 and 1.000, respectively. IndelRF was also evaluated on cancer germ line mutations; 72 insertions and 19 deletions with accuracies of 0.972 and 1.000, respectively. IndelRF outperformed the existing algorithms in these data sets (Table 2).
Prediction Accuracies Compared Between Methods for Four ClinVar Test Sets in Indels
3.4. Identifying pathogenic inframe indel mutations in MOKCa
We applied IndelRF to the inframe indels identified in the MOKCa database. A total of 844 unique insertion and 1790 deletion mutations were identified. Of these (46%), 392 insertions were predicted to be pathogenic in 251 genes, and 848 (47%) deletions across 611 genes. Based on the cancer gene classification in the Cancer Gene Census (Futreal et al., 2004), we then identified which of these mutations occurred in oncogenes as tumor suppressors. A set of 98 deletions in 37 oncogenes and 134 deletions across 31 tumor suppressors were predicted to be pathogenic deletions (Supplementary Tables S10 and S11). This suggests that deletions can be both activating in oncogenes and causing gene disruption in tumor suppressors. We also detected 80 putative activating insertions across 26 oncogenes and 69 inactivating insertions across 18 tumor suppressors (Supplementary Tables S12 and S13).
Below are some of the indels predicted to be pathogenic, confirmed by reports in the literature.
3.4.1. EGFR p.L747_E749delLRE
Epidermal growth factor receptor (EGFR) is an oncogene that regulates cell proliferation. Mutations in EGFR activate the EGFR signaling pathway and promote EGFR-mediated prosurvival and antiapoptotic signals through downstream targets such as RAS, RAF, and MEK (Zhang et al., 2010). The most abundant EGFR mutations are deletions in the kinase domain in exon 19 (residues 747–752) and constitute about 45% of all EGFR mutations (Zhang et al., 2010). These mutations are thought to alter the structure of the kinase that results in the active conformation becoming more stable than the inactive conformation, and hence the kinase becomes constitutively active.
3.4.2. JAK2 p.E543_D544del
Similarly Janus kinase 2 (Jak2) is an oncogene that promotes the growth and division of cells. Jak2 mutations define a distinct myeloproliferative syndrome that affects patients with a diagnosis of polycythemia vera (PV) (Scott et al., 2007). A small faction of PV patients (<5%) carry usually deletion mutations in JAK2 at exon 12 (Cazzola and Kralovics, 2014; Tefferi and Pardanani, 2015) at residues E543 (Scott et al., 2007).
3.4.3. KRAS p.G10_A11insG
KRAS is one of the RAS superfamilies that act as oncogenes. It helps regulate cell growth. When mutated cell signaling is disrupted, it leads to uncontrolled cell proliferation and the development of cancer. KRAS insertion mutations have been observed between codons 10 and 11 (KRAS p.G10_A11insG) in one patient with colorectal cancer (Tong et al., 2014) and also in one myeloid leukemia patient (Bollag et al., 1996).
3.4.4. ARIA1A p.Q1334delQ
AT-rich interactive domain 1A (ARID1A) is a tumor suppressor that has been recognized in several types of human cancers. About 5% of ARID1A somatic mutations are inframe indels (Guan et al., 2012). In deletion mutations at position Q1334del were found two tumors, gastric carcinoma (Jones et al., 2012) and prostate carcinoma (Wang et al., 2011).
4. Conclusions
In this study, we sought to develop machine-learning models to identify pathogenic inframe indels. We compared the ability of six prediction tools to discriminate between these pathogenic mutations and a set of neutral mutations, which they all did with ease.
We then developed our own classifiers that could discriminate pathogenic mutations with an accuracy of 0.995 and 0.968 for insertions and deletions, respectively. The four most important features of our classifiers were the VEST p-value, priPhCons, Phylop, and Gerp++ of inframe insertion and deletion mutations.
Finally, we have used our algorithms to predict the functional consequence of 844 insertion mutations and 1790 deletion mutations documented in the MOKCa database.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.