
Abstract
Abstract
Duplicate sequence records—that is, records having similar or identical sequences—are a challenge in search of biological sequence databases. They significantly increase database search time and can lead to uninformative search results containing similar sequences. Sequence clustering methods have been used to address this issue to group similar sequences into clusters. These clusters form a nonredundant database consisting of representatives (one record per cluster) and members (the remaining records in a cluster). In this approach, for nonredundant database search, users search against representatives first and optionally expand search results by exploring member records from matching clusters. Existing studies used Precision and Recall to assess the search effectiveness of nonredundant databases. However, the use of Precision and Recall does not model user behavior in practice and thus may not reflect practical search effectiveness. In this study, we first propose innovative evaluation metrics to measure search effectiveness. The findings are that (1) the Precision of expanded sets is consistently lower than that of representatives, with a decrease up to 7% at top ranks; and (2) Recall is uninformative because, for most queries, expanded sets return more records than does search of the original unclustered databases. Motivated by these findings, we propose a solution that returns a user-specified proportion of top similar records, modeled by a ranking function that aggregates sequence and annotation similarities. In experiments undertaken on UniProtKB/Swiss-Prot, the largest expert-curated protein database, we show that our method dramatically reduces the number of returned sequences, increases Precision by 3%, and does not impact effective search time.
1. Introduction
B
A key challenge to the quality and efficiency of database search is duplication, that is, the presence of multiple records that contain similar or even identical sequences (Bursteinas et al., 2016). Duplication has two immediate impacts on database search: it dramatically increases database search time and the top-ranked retrieved sequences can be highly similar. Together, these impacts lead to uninformative search results and make it difficult to find potentially interesting sequences that are only slightly less similar to a search query. A possible solution is to remove duplicate records. However, the notion of duplication is context-dependent; removal of records that might be regarded as duplicates for one task may be harmful to other tasks (Chen et al., 2017b).
Machine learning techniques are often used to solve computational problems in biology, such as clustering methods that have been widely applied (Fu et al., 2012) to address duplication. These methods cluster a sequence database at a user-defined sequence identity threshold, creating a non-redundant database. Users search against representative records (one sequence per cluster) of nonredundant databases and can further expand search results by exploring records from the same clusters. Search against representatives reduces search time and provides more diverse search results; exploration of cluster members, in addition, should ensure that potentially interesting records will still be found.
To assess the search effectiveness of nonredundant databases, Precision and Recall have been used in existing studies (Suzek et al., 2015). BLAST all-by-all search results (where each sequence was compared with all sequences in a database via BLAST) from original databases are used as a gold standard. Precision measures the proportion of retrieved sequences labeled as relevant, based on this gold standard, out of the total number of retrieved sequences from nonredundant databases; Recall measures the proportion of relevant sequences retrieved by nonredundant databases out of the total number of relevant sequences in the gold standard set. Nevertheless, the use of Precision and Recall has limitations. Precision assumes that users will examine all the retrieved sequences; however, this is not realistic: biological database users pay more attention to top-ranked retrieved sequences (Cole et al., 2008). Likewise, Recall is not an effective information retrieval measurement, evident by findings in related studies for over a decade (Zobel et al., 2009; Webber, 2010; Walters, 2016).
In this study, we focus on search effectiveness in nonredundant databases. * The contributions are twofold. First, we propose metrics to better model user behavior and reassess search effectiveness, summarized in Section 5. The evaluation results demonstrate that Recall is not effective in information retrieval tasks. For example, reaching Recall of more than 90% is at the cost of users having to manually examine more nonrelevant sequence records for about 90% of queries than in original databases. Furthermore, the results illustrate that the Precision of expanded clusters (representatives and members) is distinctly lower than the Precision when only using representatives. Second, to address these problems, we propose a ranking model that returns a user-specified proportion of top similar cluster members, based on their sequence and annotation similarity to representatives, summarized in Section 6. We demonstrate that it improves the Precision of expanded clusters, facilitates more satisfactory user search by providing only top similar cluster members instead of all results, and does not increase practical searching time.
2. Duplication in Biological Sequence Databases
Biological sequence databases archive knowledge about sequences, structures, and biological function. They are used for data storage: newly sequenced records are submitted to databases and corresponding database record IDs are cited for research communications (Karsch-Mizrachi et al., 2017). They are also used for data search: uncharacterized sequences are searched against database records for homology prediction, coding region identification, and function classification (Gish and States, 1993).
Advances in high-throughput sequencing mean that, on the one hand, genomes are being sequenced ever more efficiently with fewer errors. On the other hand, this has led to a vast increase in the number of sequence records deposited in databases. For instance, GenBank (a primary nucleotide database coordinated by the National Center for Biotechnology Information) has more than 200 million nucleotide records consisting of more than 250 billion base pairs. Its average annual growth in terms of the number of base pairs was 35.52% (Benson et al., 2017). Likewise, UniProtKB/TrEMBL (a primary protein database coordinated by the UniProt Consortium) contains more than 120 million protein records, almost doubling in size in 2016 (The UniProt Consortium, 2018).
Such dramatic increases in numbers of sequence records lead to duplication. Duplicate records can be broadly categorized as of two kinds: entity duplicates, which are records belonging to same entities (Chen et al., 2016b, 2017b; Yonchev et al., 2018), such as when the same gene records are submitted to the same database (Chen et al., 2017b); and near-duplicates or redundant records (Suzek et al., 2015; Mirdita et al., 2016; Chen et al., 2018), where records share some specified percentage X% similarity defined by users. For example, the Uniclust protein database defines redundant records at sequence similarity thresholds of 30%, 50%, and 90% for different purposes (Mirdita et al., 2016). Both kinds of duplicate records have impacts on different use cases. In biocuration (biological data curation), biocurators have to manually resolve conflicts and inconsistencies caused by entity duplicates (Magrane and UniProt Consortium, 2011); in database search, users have to manually explore repetitive or near identical search results caused by redundant records (Bursteinas et al., 2016). We focus on redundant records in this article.
Redundant records challenge database search in terms of both efficiency and effectiveness. A particular instance was the high level of redundancy in UniProtKB/TrEMBL. Records from different strains of the same bacterial species were deposited; for example, records from 1692 strains of Mycobacterium tuberculosis were overrepresented in close to 6 million records. † Such redundancy dramatically lowers the search speed and brings highly repetitive search results. To address this, UniProt staff removed about 50 million redundant records using a combination of manual and automatic approaches (Bursteinas et al., 2016). Similar examples were also observed in sequencing repositories, where database managers encourage a joint effort to handle redundancies (Gabdank et al., 2018).
3. Sequence Clustering to Address Redundancies
Clustering is the main technique currently used to address redundancies in biological sequence databases. It is an unsupervised machine learning approach that groups records based on a similarity function. We have detailed sequence clustering methods and their related applications in previous works (Chen et al., 2016a, 2018). Briefly, sequence clustering methods are greedy algorithms that aim to reduce the number of sequence alignments. It has the following three primary steps (Fu et al., 2012):
Sequences are sorted by decreasing length. The longest sequence is by default the representative of the first cluster. The remaining sequences are processed in order. Each is compared with the cluster representative. If the sequence identity for some cluster is no less than the user-defined threshold, it is assigned to that cluster as a member. It will be a new cluster representative if it is not similar to any existing representatives. The generated representatives and corresponding members comprise the nonredundant (or clustered) database.
3.1. Sequence search on nonredundant databases
Figure 1 compares sequence search on the UniProtKB database, consisting of UniProtKB/Swiss-Prot and UniProtKB/TrEMBL records, and UniRef50, a nonredundant database created by clustering at a threshold of 50% similarity from UniProt records. Sequence search on a common database compares query sequences against all the database records (assuming that no heuristics are applied). In some cases, it produces highly similar search results due to redundancies, as shown in Figure 1a.
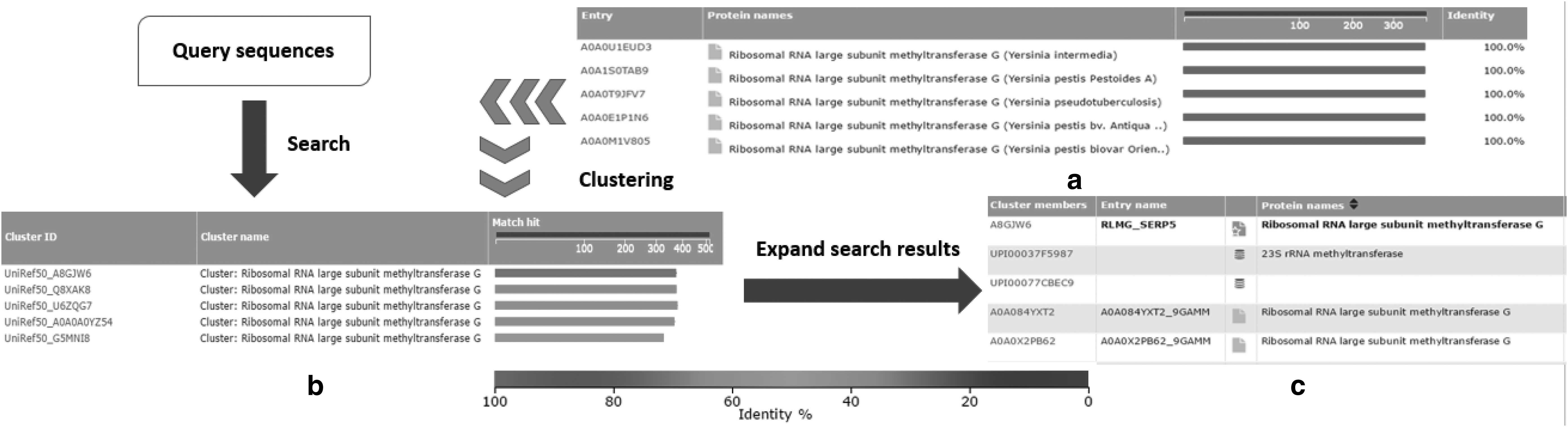
Search of query sequences against original database versus nonredundant database using search results of UniProtKB/Swiss-Prot record A7FE15 on UniProtKB and UniRef50 (a clustered database) as an example.
In contrast, sequence search on nonredundant databases consists of two steps. First, query sequences are only searched against cluster representatives, as shown in Figure 1b. The retrieved records are effectively a ranked list of cluster representatives in the nonredundant database. Second, search results are further expanded by exploring associated cluster members if users are interested in the retrieved representatives, as shown in Figure 1c. By searching against representatives only, the search takes less time and brings more diverse results. By expanding the clusters, users can explore the members that are similar to cluster representatives to confirm their findings.
4. Existing Search Effectiveness Measures and Limitations
Previous studies used Precision and Recall to assess the search effectiveness of nonredundant databases (Suzek et al., 2015; Mirdita et al., 2016). The formulas are shown in Equation (1).
Here, we explain how UniRef staff used Precision and Recall to evaluate search effectiveness as an example (Suzek et al., 2015). First, a BLAST all-by-all search was performed on the UniProtKB database. The BLAST results are commonly called query–target pairs or hits, where a query sequence is paired with multiple retrieved sequences in a database ordered by BLAST scores. The query–target pairs with BLAST e-value no more than a user-defined threshold, such as 10, were selected as gold standard relevant hits, identified as G in the equation. Another BLAST all-by-all search was performed on UniRef50 database. ‡ The expanded query–target pairs—the representatives satisfying the e-value threshold and their cluster members—were considered as retrieved results (identified as E in the equation). Precision measures the proportion of relevant results out of all retrieved results: given E retrieved target-pairs, the number identified as relevant in the gold standard. Recall measures the proportion of relevant results out of total relevant query–target pairs: given G relevant target-pairs, the number of them retrieved from the nonredundant database. The evaluation results on UniRef50 illustrate that Precision and Recall were consistently more than 0.83 and 0.95, respectively, for multiple thresholds.
Precision and Recall are popular metrics for the measurement of classification tasks: in this case, Precision quantifies the accuracy of search results on nonredundant databases; Recall quantifies whether important search results can be retrieved via nonredundant databases. However, the adopted metrics and evaluations do not reflect user behavior in practice. Specifically, there are three limitations of concern.
First, users may not expand cluster members when searching nonredundant databases. Some use cases have a higher priority on search diversity, where only representatives are used as search results. For instance, Malde and Furmanek (2013) only used representatives of UniRef50 to find a large diversity of protein sequences for identification of protein structures via transitive alignments. Similarly, Gu et al. (2016) only used representatives of UniRef90 for sequence evolutionary conservation analysis. In contrast, expansion of cluster members is more important for other cases. For example, Remita et al. (2016) searched against UniRef for miRNAs regulating glutathione S-transferases and expanded the results from the associated UniRef clusters to obtain alignment information, Gene Ontology (GO) annotations, and expression details to ensure that they did not miss any other related data (Remita et al., 2016). Thus, the evaluation should consider both circumstances.
Second, the relevance of top-ranked results can be critical to users. Precision assumes that users will examine all the retrieved sequences and judge whether they are of interest. Nevertheless, this assumption is not realistic in practice. For large collections such as UniProt databases, a BLAST search could yield over hundreds of hits. It is tedious for users to manually check all the hits, and it is not necessary because only top-ranked hits have high BLAST alignment scores (or, equivalently, lower e-values). For instance, Cole et al. (2018) created a protein sequence structure prediction website that searches user-submitted sequences against UniRef and selects the top retrieved representatives based on e-values. Thus, the Precision should be adapted to focus more on top-ranked results.
Third, Recall also has failings. It has been a long-term concern that Recall may not be effective for information retrieval measurement because it is not unrelated to user satisfaction level in regard to any single query (Zobel, 1998; Webber, 2010; Walters, 2016). As we will quantitatively show later, a high Recall means that users have to manually examine more nonrelevant hits.
5. Reassessing Search Effectiveness
We propose updated evaluation metrics and reassess the search effectiveness of nonredundant databases, as summarized below.
5.1. Proposed evaluation metrics
Enhanced evaluation metrics are proposed for modeling user behaviors, as follows:
The denotations are as follows. Given a query Q, let F be the list of fetched (retrieved) representatives from the nonredundant database, E its expanded set, and R the set of relevant sequences. Here, F is a ranked list, consisting of representatives ordered by BLAST scores, whereas E contains representatives and the associated cluster members, which do not have a particular order. R in this case stands for all the fetched sequences for Q from the original databases as the gold standard. Each sequence, either in F or in E, has a relevance score based on a scoring function S, that is, 0 if it is not in R, or 1 otherwise.
As previously mentioned, the Precision of representatives only (F) and expanded clusters (E) should be evaluated separately. For representatives only, the existing metric
5.2. Data set, tools, and experiments
We used the complete UniProtKB/Swiss-Prot Release 2016–15 as our experimental data set. It consists of 551,193 protein sequence records. CD-HIT (4.6.5) was used to construct the associated nonredundant UniProtKB/Swiss-Prot; NCBI BLAST (2.3.0+) was used to perform all-by-all searches. UniProtKB/Swiss-Prot is the largest expertly curated protein databases; CD-HIT is one of the most popular sequence clustering tools. They were selected as representatives.
CD-HIT by default removes sequences of length no greater than 10 since such short sequences are generally not informative. We removed those records correspondingly in the complete UniProtKB/Swiss-Prot. The updated data set has 550,047 sequences. We used them as queries and performed BLAST searches on the updated UniProtKB/Swiss-Prot and its nonredundant version at 50% threshold generated by CD-HIT. The nonredundant database at 50% consists of 120,043 sequences. 547,476 of 550,047 query sequences have at least one retrieved sequence in both databases. The commands for running CD-HIT § and BLAST ** strictly follow user guidance. NCBI BLAST staff (personal communication via e-mail) advised on the maximum number of output sequences to ensure sensible results. Note also that this study focuses on general uses of the tools, while, for instance, UniRef and Uniclust may use different parameters to construct nonredundant databases for specific purposes.
For BLAST query–target pairs obtained by all-by-all searches, we removed two types of query–target pairs: where the target is the query itself; and the same sequence retrieved more than once for a query. BLAST performs local alignment—a target sequence may be retrieved multiple times for the same query sequence if its different regions (subsequences) are similar to the query. In this case, it biases statistical analysis.
We measured the Precision for both representatives (
5.3. Search effectiveness results and discussion
Our search effectiveness results illustrate two main observations.
The Precision of the expanded set distinctly degrades at top-ranked hits. Table 1 summarizes different levels of
Representatives:
Recall is overestimated and in turn is not informative, due to the expanded set having even more query–target pairs than the original data set. Figure 2a compares the number of query–target pairs. The retrieved pairs among representatives include only about 15% of the pairs from the original data set. On the one hand, this indicates that nonredundant databases dramatically reduce search time and users can browse the search results more efficiently. On the other hand, it shows that expansion of results is valuable since potential interesting records may be in the other 85%. However, the expanded set produces 40,095,619 more pairs than the original. Figure 2b further shows that the expanded set produces more pairs on over 89% of queries (492,129 of 547,476), and on average produces about 10 pairs per query (Fig. 2c). Having more pairs results in high Recall. Both median and mean Recall (Fig. 2d) are above 90%, but this comes at the cost of producing more than 40 million pairs. We further computed Jaccard similarity (the number of shared query–target pairs over the total number of pairs between E and R). Jaccard similarity by comparison is almost 20% lower than Recall, which clearly shows the results of the expanded set are not similar to those of the original database. This observation is consistent with study results reported from other domains, showing that Recall is not an appropriate metric for search effectiveness measurement.
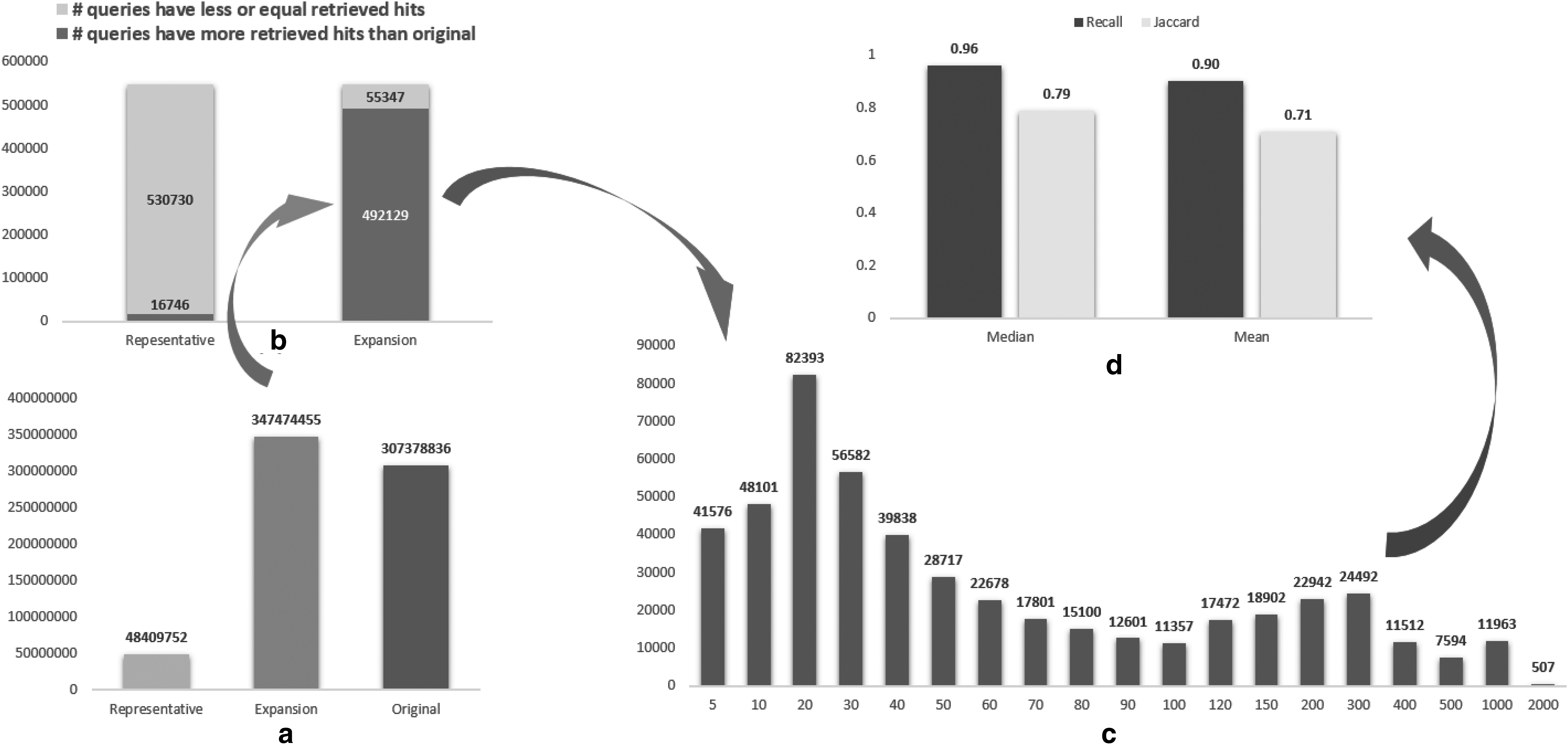
6. Improving the Search Effectiveness of Expanded Clusters
Following these observations, we propose a solution that ranks cluster members in terms of their similarity to cluster representatives and only returns the top X%, a user-defined proportion, when they expand search results. To the best of our knowledge, existing databases such as UniRef select representatives based on whether a record is reviewed by biocurators, is from a model organism, or other such record-external factors. They do not compare and rank the similarity between records. Also, they expand all the records in a cluster rather than choosing only a subset.
6.1. A ranking model
In our proposal, the notion of similarity between a record and its cluster representative is modeled based on sequence identity and annotation similarity. This similarity function is shown in Equation (5), where R and M refer to a representative and an associated cluster member record, respectively.
The records in each cluster are thus ranked by this similarity function in descending order. The top-ranked X% records, with parameter X specified by a user, will be presented when the user expands search results. The ranked model can be adjusted by both database staff and database users. On the one hand, database staff can customize the ranking function, such as adjusting weights and selecting different types of annotations, when creating nonredundant databases. On the other hand, database users can select how many records to browse rather than seeing all records when expanding search results.
We used sequence identity reported by CD-HIT and molecular function (MF) GO term similarities as annotation similarity. MF GO terms are extracted from the UniProt-GOA data set (Courtot et al., 2015), and the similarity is calculated using the well-known LinAVG metric (Lin, 1998). We applied the ranking function with two sets of weights: the first is when
6.2. Ranking model results and discussions
Table 1 compares detailed
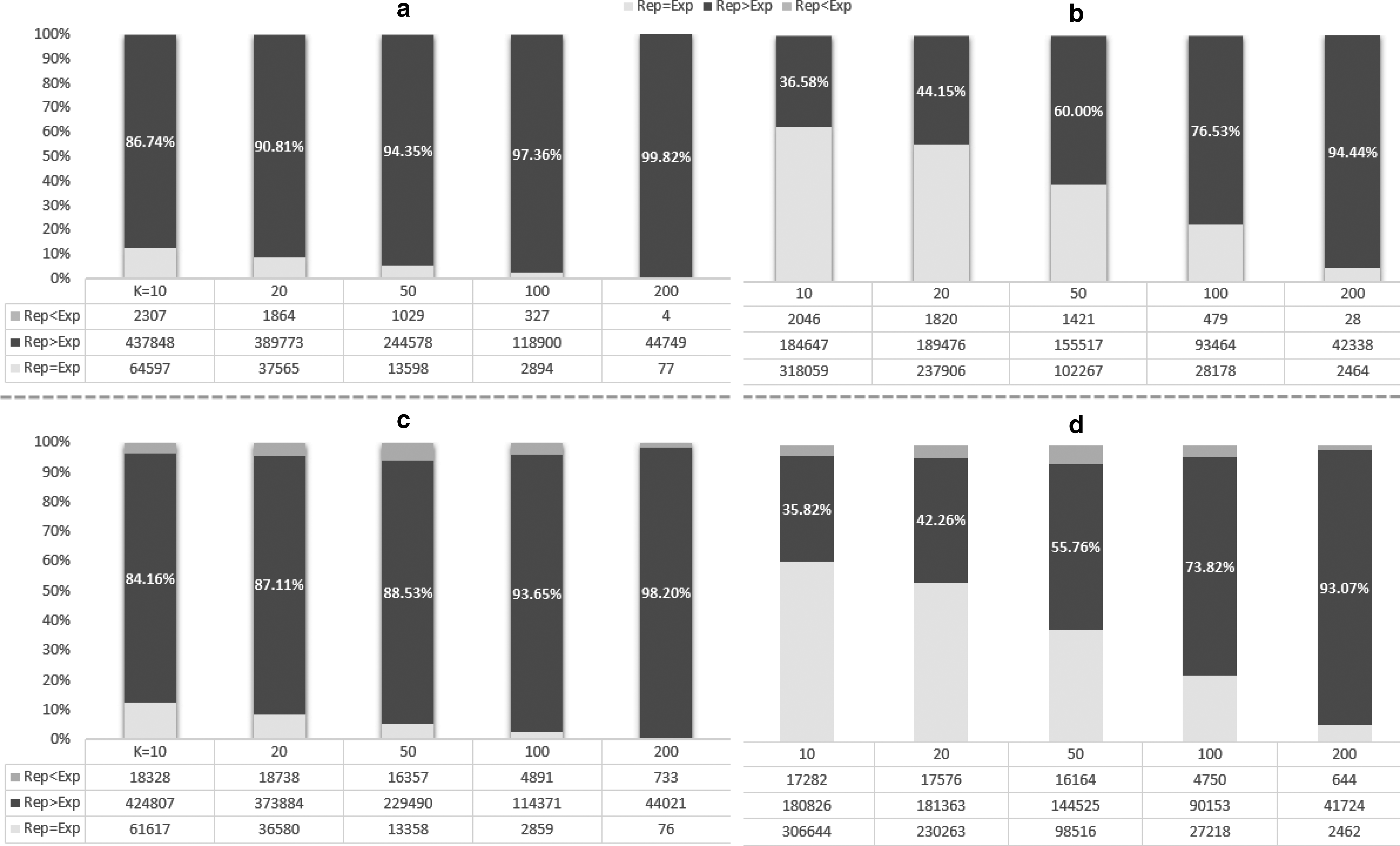
Proportion of queries having higher Precision in representatives than in the expanded set. We removed queries that have the same number of hits in both (it means retrieved representatives do not have any member records). The first row compares unranked expanded set
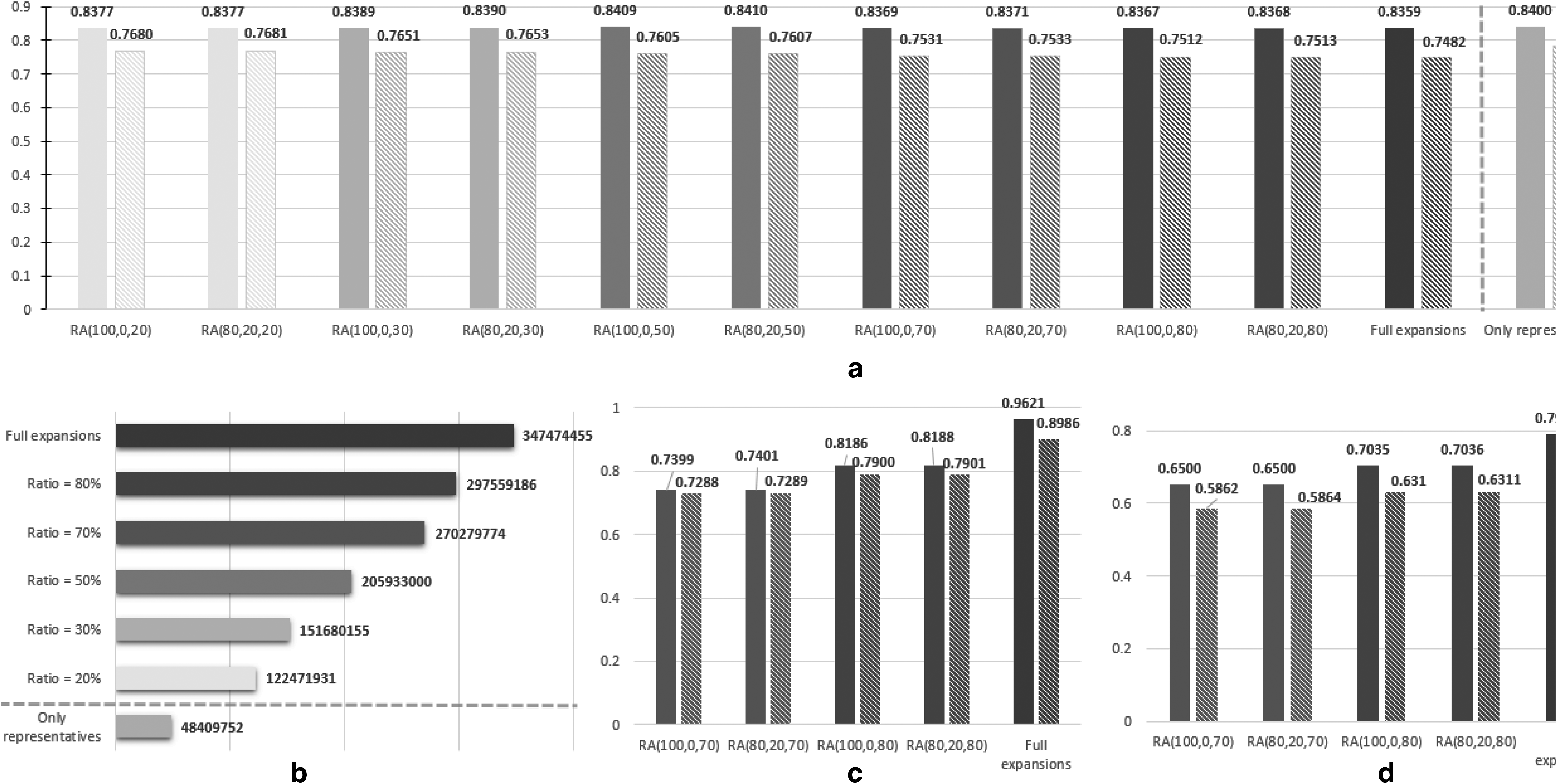
Comparative results for original (unranked) expanded set and our proposed ranked model. Subgraphs
A limitation of the approach is that it has lower Recall than the full expanded set (shown in Fig. 4c, d). However, it is our view that the number of expanded query–target pairs and Precision measures are more critical to user satisfaction. For instance, proportion at 20% produces around 200 million fewer query–target pairs and has 2% higher
7. Conclusion
We analyzed the search effectiveness of nonredundant databases generated by sequence clustering methods. We propose innovative evaluation metrics to better model user behaviors. The detailed assessment results illustrate that the Precision of representatives is high, but that expansion of search results can degrade Precision and reduce user satisfaction by producing large numbers of additional hits. We propose a model that ranks cluster members in terms of sequence identity and annotation similarity. The comparative results show that it has the potential to bring more precise results and reduce user effort, that is, yielding efficient and accurate discovery of relevant answers.
Footnotes
Acknowledgments
We appreciate the advice of the NCBI BLAST team on BLAST-related commands and parameters. The work of Q.C. is supported by the Melbourne International Research Scholarship from the University of Melbourne. The project receives funding from the Australian Research Council through a Discovery Project grant, DP150101550.
Authors' Contributions
Q.C. and X.Z. conceived and designed the experiments. Q.C. performed the experiments. All the authors analyzed the data. Q.C., J.Z., and K.V. wrote the article. All the authors approved the final article.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
‡
Note that UniRef50 contains records from sources other than UniProtKB; only overlapped sequences were selected for evaluation.
§
./cd-hit -i input_path -o output_path -c 0.5 -n 2, where -i and -o stand for input and output path. -c stands for identity threshold and -n specifies word size recommended in the user guide.
**
./blastp -task blastp -query query_path -db database_path -max_target_seqs 100000, where blastp specifies protein sequence, -query and -db specifies query and database path. -max_target_seqs is the maximum number of returned sequences for a query.