
Abstract
Abstract
1. Introduction
Benchtop genome sequencers such as the Illumina MiSeq, MiniSeq, or iSeq (https://www.illumina.com) are revolutionizing genomics research for smaller independent laboratories, by enabling low-cost access to next-generation sequencing (NGS) technology. However, postsequencing bioinformatics data analysis still presents a significant bottleneck for smaller laboratories or clinical facilities lacking bioinformatics expertise. Although user-friendly bioinformatics cloud computing solutions aim to address this challenge (Krampis et al., 2012; Krampis and Wultsch, 2015), they can be prohibitive due to recurring high costs. For example, Illumina BaseSpace (https://basespace.illumina.com) offers an integrated solution where the NGS data are streamed directly from the sequencer to the cloud, where users can access a range of bioinformatics applications through an intuitive web interface. However, BaseSpace is costly solution as it requires a yearly subscription of $4,999 and additional “iCredit” payments for running the applications. In contrast, the cost of computer hardware in recent years has greatly declined, and an Intel Xeon server with 64 Gigabyte (GB) memory and multiple Terabyte (TB) of storage, for example, costs less than the yearly subscription to BaseSpace. Therefore, although hardware with sufficient computational capacity to analyze data from small factor NGS instruments is well within the reach of independent laboratories, data analysis is still a significant bottleneck due to the lack of easily accessible bioinformatics solutions for nonexperts.
2. Implementation
We have developed miCloud, a bioinformatics platform that leverages cloud technologies for providing an easy-to-use streamlined NGS data analysis using local computational hardware. Laboratories lacking bioinformatics experts on their team can easily process genomic data generated using in-house sequencing instruments or outside providers, through the intuitive user interface dashboard (Fig. 1i). Furthermore, the dashboard allows users to import and manage their NGS data through a visual file explorer (Supplementary Material), and perform data analysis through a range of NGS data pipelines, with the default or by changing the algorithm parameters (Fig. 1ii). Parallel data processing can also be performed by running multiple instances of each pipeline, and users can monitor the status and progress of each run on the dashboard.
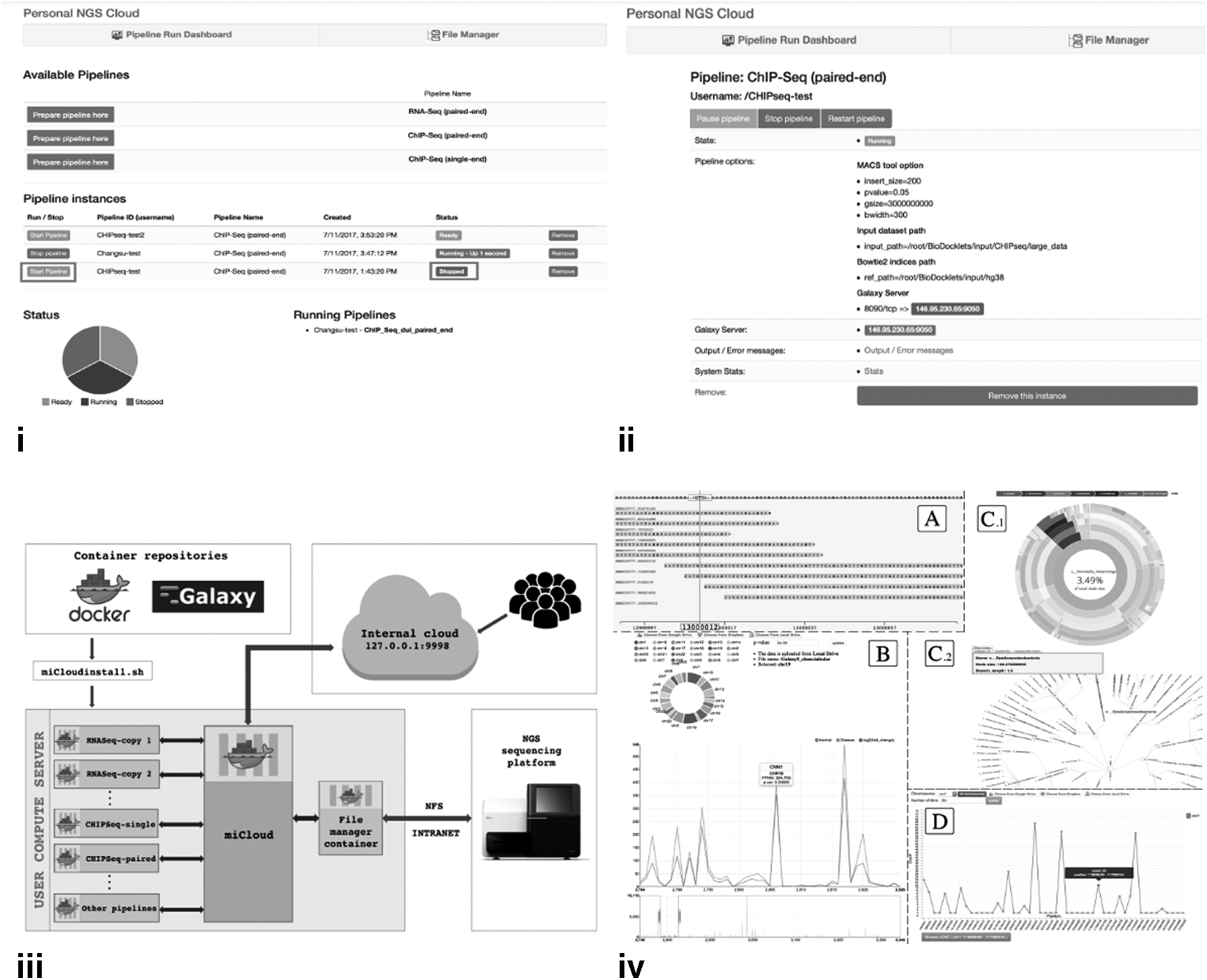
The miCloud is highly modular since it is implemented with Docker technology (www.docker.com) at its foundation, whereas it encapsulates each different component (e.g., user interface, file manager, and data analysis pipelines) in a different container (Fig. 1iii). The containers are automatically instantiated and interconnected upon installation, which is performed by running a script available on our code repository (https://github.com/BCIL/Personal-NGS-Cloud). The miCloud can be deployed on a computer server, university cluster, or desktop computer, and connected to a genome sequencing instrument for direct data transfer over the local network, through the Network File System (NFS) protocol that is built in the Docker containers (Supplementary Material).
By default, the miCloud includes three preconfigured and ready-to-execute NGS pipelines: two for single or paired-end ChIP-Seq data and one for paired-end RNA-Seq data (Supplementary Material). Each pipeline provides beginning-to-end bioinformatics analysis following the standard published protocols for RNA-Seq and CHIP-Seq. If required, researchers can easily modify these pipelines through the Galaxy workflow canvas that is also preconfigured in the containers (https://galaxyproject.org/learn/advanced-workflow/basic-editing/) and accessible through a link on the miCloud dashboard (Supplementary Material). Furthermore, we have integrated in each container our previously published Visual Omics Explorer framework, providing rich interactive Javascript-D3 (https://d3js.org/) visualizations and publication-ready graphics in the miCloud file outputs. Example visualizations include those for NGS read alignments, RNA-Seq differential gene expression, and display of CHIP-Seq peaks on a genomic region (Fig. 1iv(A–D), respectively).
Our implementation follows the same principles previously published on the Bio-Docklets project (Kim et al., 2017), using the BioBlend software library (https://bioblend.readthedocs.io/en/latest/; Sloggett et al., 2013) for controlling pipeline execution on the back end of the Galaxy server (Afgan et al., 2018). Similarly, for the miCloud platform we have developed a set of custom BioBlend scripts (https://github.com/BCIL/Personal-NGS-Cloud), which translate data operations performed by the users on the graphical dashboard (Fig. 1i,ii) to commands issued to the Galaxy Application Programming Interface (API; https://galaxyproject.org/develop/api/). The Galaxy API is fully encapsulated inside the miCloud Docker containers, which in turn abstracts the details of the pipeline execution and provide easy access for nonexpert users to run complex bioinformatics data analysis through the miCloud dashboard. Furthermore, users can also manage raw NGS read data and outputs of the pipeline runs through the dashboard, which are located on the server running miCloud, or on a genome sequencing instrument that is accessible through NFS over the local network.
The miCloud software can easily be deployed by running a single script, available along with all dependencies in a .zip file, on our code repository (https://github.com/BCIL/Personal-NGS-Cloud). The script automatically performs the miCloud deployment, guiding users through all steps and the required parameters such as installation directories or permissions. In addition, we have prepared online video tutorials demonstrating the process (Supplementary Material). Upon completion of the installation, an Internet protocol (IP) address for the container with the dashboard is printed by the script, which can be simply entered by the users on a web browser to access the miCloud interface and start an NGS data analysis run.
3. Discussion
The miCloud provides researchers with a user-friendly fully extensible bioinformatics platform for analysis of large-scale genome sequencing data sets, without requirement of any prior bioinformatics expertise. A set of preconfigured pipelines enables users to efficiently process NGS data generated in their laboratory or at external sequencing providers and produce rich interactive visualizations that can be exported as high-quality graphics. Furthermore, the miCloud implementation fully abstracts the underlying bioinformatics software complexity by encapsulating the data pipelines within Docker containers and the standardized Galaxy API. Users can seamlessly integrate a genome sequencer with a fully featured on-site data analysis cloud and remove the data analysis bottleneck to integrate NGS as part of the standard research protocols in a biological sciences or medical laboratory. With the availability of low-cost benchtop sequencing instruments, the miCloud platform can democratize NGS bioinformatics for smaller independent laboratories, whereas at the same time simplify genomic data analysis similarly to the way reagent kits for biological sample preparation have streamlined laboratory operations. Our implementation also follows the principles of standardization set forth by the BioCompute Objects (Simonyan et al., 2017; Alterovitz et al., 2018) toward standardized workflow access and interoperability across the industry, scientists, regulators, and other stakeholders in biocomputing.
The technologies we selected as the foundation for implementing the miCloud enabled us to provide a fully extensible and adjustable solution where users can easily integrate new, or modify the existing bioinformatics pipelines available on this platform. Specifically, the IP address of each Docker container is shown on the miCloud dashboard, and by entering this address on a web browser users can access the Galaxy workflow canvas (https://galaxyproject.org/learn/advanced-workflow/basic-editing/) running inside our containers (Supplementary Material). Our preconfigured pipelines can then be modified easily by a researcher through the canvas, without any requirement for command-line expertise. Furthermore, new pipelines can be implemented, and users can install additional bioinformatics algorithms with a single click through the Galaxy ToolShed (Blankenberg et al., 2014).
Developers can utilize the miCloud platform for providing access to a large selection of ready-to-execute bioinformatics pipelines, available from online Docker repositories such as BioContainers, DockStore, or BioShadock (http://biocontainers.pro, http://bioshadock.genouest.org, https://dockstore.org/). Although tools on these repositories come preconfigured within Docker, for their majority they are accessible only through a command-line login to the containers. Bioinformatics developers can leverage our platform to make these tools easily accessible for nonexperts, through the miCloud dashboard and file manager interface. Furthermore, beyond small laboratories the miCloud can serve the needs of large core facilities as it can be similarly deployed on a university computer cluster, where it can be used as a central platform for processing the genomic data for tens or hundreds of samples in large-scale NGS studies.
Footnotes
Acknowledgments
Funding was done by National Institute on Minority Health and Health Disparities (G12 MD007599) and Weill Cornell–CTSC (2UL1TR000457-06).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.