
Abstract
Abstract
Massively parallel sequencing (MPS) has become a standard technique in molecular biology whose application has spread from the analysis of the human genome to that of virtually all other organisms. MPS requires reference genomes to be performed and, in some cases, multiple genomes need to be handled as a single unit to carry out genetic analysis. Nucleic acid sequences are typically stored in “fasta” files, which can contain multiple genomes (“multi-fasta”). Although it is possible to convert a multi-fasta file into a single sequence using specific computer commands, the resulting file will not keep track of the boundaries of the original sequences, making it difficult to determine to what genome read obtained from MPS belong to. In this study we introduce mingle, a shell script that can be used to create custom reference genome by merging multi-fasta files while providing a list of boundaries of the individual genomes that can be used for downstream analysis.
1. Introduction
Massively parallel sequencing (MPS), a technique that has uncovered the complexity of the human genome (Clarke et al., 2012), has become a standard procedure in the laboratory of molecular biology that is providing valuable information related to the genomic structure and genetic expression of microorganisms (Tagini and Greub, 2017; Tang and Larsson, 2017). Apart from de novo genome assembly experiments, MPS requires a short read aligner to map the sequences obtained during the experiment to a known reference genome (Ruffalo et al., 2011; Pfeifer, 2017).
Although there are plenty of short read aligners, there are few available reference genomes limited to the most common organisms; for instance, the integrative genome viewer (IGV) (Robinson et al., 2011), which can graphically display the obtained MPS reads, is supplied with a set of 79 unique genomes belonging to 58 families of organisms, a tiny fraction of the more than three trillion sequences deposited into the GenBank (Sayers et al., 2019). The availability of reference genomes represents a bottleneck in metagenomics, for less popular reference genomes need to be prepared by the operator to perform MPS analysis.
Although it is possible to download reference sequences from the GenBank by different approaches, there are situations where it is not feasible to work with single reference sequences. For instance, it might not be known beforehand what exact organism the reads under investigation belong to, or there could be occurrences of recombination between multiple genomes. In these cases, a panel of reference sequences should be used as a single unit.
The most common file format for nucleic acids and amino acid sequences is the fasta format, which allows multiple entries on the same file (multi-fasta). In this format, each sequence contains a header introduced by the character “>” and terminated by a new line; the actual sequence is then spread into one or more lines each typically having the same number of characters (line width), usually 80 (Zhang, 2016). To restate the process of merging a multigenomic reference file using the fasta format, the resulting file should have a single header and a cluster of nucleotides derived from all the individual genomes. It is possible to perform this action with, for instance, Unix/Linux shell commands (such as: grep -v "^>" file.fa > newfile.fa). However, the individual sequences might have a variable line width that would be passed to the processed file. In addition, using this approach, it is no longer possible to find the boundaries of the individual genomes.
There are some software focused on merging fasta files. For instance, MultiFASTA Builder (www.dnabaser.com/help/tools-converters/MultiFASTA%20Builder/index.html) is a proprietary program for the Windows operative system that takes as an input a list of single fasta files and then creates a multi-fasta. The Sequence Manipulation Suite (Stothard, 2000) is a freeware program also available as an online platform (www.bioinformatics.org/sms2/combine_fasta.html) that does perform merging but does not show the boundaries of the original sequences.
In this study we introduce mingle, a utility for merging multi-fasta files that allows the definition of the output's line width and that provides the boundaries of the individual sequences for downstream applications. Mingle also performs a test on the length of the individual sequences to prove that the boundaries were properly determined.
2. Materials, Methodologies, and Techniques
Mingle version 2.3 was named after the verb “to mingle” meaning to blend, to mix, to coalesce; it was written in Bash language and tested on the Linux distributions Mint 20, openSUSE 13, and Ubuntu 18.04 running Bash version 4.4.19, 4.2.53, and 4.4.19, respectively. In this study, the path to the mingle executable was provided in the Linux environmental variables, thus it was possible to run mingle as any other shell commands simply by typing mingle <options> on a Linux terminal; alternatively, mingle could be launched as a common bash scripts. Mingle's script is given in Supplementary File S1.
Mingle has a manual page that can be displayed with the command: mingle -h. Mingle works following the syntax: mingle <input> <output> <header>, where input is the name of a multi-fasta file (including the extension; this approach was chosen because it was assumed that the user would use any kind of extension or none at all), output provides the name of the final files (one with the extension “.fa” containing the merged sequences and one with the extension “.tsv” containing the boundaries of the individual sequences), and header represents the name of the fasta header (if this is formed by multiple words, it should be provided within quotes). A restriction of mingle is that the headers of the individual sequences should be unique.
Following the “rule of silence” of the Unix philosophy (https://en.wikipedia.org/wiki/Unix_philosophy), the execution of mingle does not show any messages to the standard output; the output of the program is entirely directed to two textual files that represent the end product of the process. To assure that mingle would not stall when dealing with large files, a progression bar could be activated with the flag -i.
Mingle calculates the cumulative length of the provided sequences to determine their boundaries; to assure that the reported lengths are correct, a test can be activated with the flag -t. With this option, mingle will use the EMBOSS (Rice et al., 2000) esearch and efetch commands to extract the GenBank file corresponding to each individual sequence. To work, this option requires a running Internet connection, the EMBOSS executable set in the environment variables, and the header of the sequences formatted according to the EMBOSS specifications, that is: >IDName An Informative comment (http://emboss.open-bio.org/html/use/ch05s02.html). Mingle will use the “IDName” field to fetch the deposited length of the genome.
Mingle can also set the width of the sequence. By default, the number of characters in each row is 100; this value was chosen because it makes easier to calculate the position of a given nucleotide when manually parsing the output file. It is possible to set the width to the more canonical 80 characters with the option -w; alternatively, a custom value can be set with the option -W n, where n is an integer. A file with a heterogeneous line width was prepared by pasting random viroid nucleic acid sequences into a text file called “unevenFile.fa”. A file with amino acid sequences was prepared by pasting the transposase sequence of Aeromonas salmonicida subsp. salmonicida culture-collection (Trudel et al., 2013) into a file called “Tn.fa”.
The alignment of selected sequences was carried out with MAFFT (Katoh et al., 2002) and the visualization of the resulting alignment was obtained with Bioedit (www.mbio.ncsu.edu/bioedit/bioedit.html).
A Bash script (“tester.sh”) was prepared to benchmark the speed of retrieving information from mingle's output as compared with enquiring remote servers (Supplementary File S2).
3. Results
Mingle was designed with the aim of full automation of the merging process. To test this design, all the viroid sequences deposited on the National Center for Biotechnology Information (NCBI) database were extracted using the EMBOSS package. The viroid taxon was chosen because it could be accessed using an identification number and because it contained enough deposited sequences to provide a multi-fasta file without requiring excessive computational resources. Retrieval and counting of the instances obtained was performed with the commands:
47
where “$” represents the prompt of a Linux terminal and 47 is the number of individual sequences retrieved. Mingle was then launched with the default line width of 100 characters; the original file “viroid.fa” was compared with the output file “mingled.fa”:
>NC_039241.1 Eggplant latent viroid complete genome, isolate 2
GGGTGGTGTGTGCCACCCCTGATGAGACCGAAAGGTCGAAATGGGGTTTCGCCATGGGTCGGGACTTTAA
ATTCGGAGGATTCGTCCTTTAAACGTTCCTCCAAGAGTCCCTTCCCCAAACCCTTACTTTGTAAGTGTGG
TTCGGCGAATGTACCGTTTCGTCCTTTCGGACTCATCAGGGAAAGTACACACTTTCCGACGGTGGGTTCG
TCGACACCTCTCCCCCTCCCAGGTACTATCCCCTTTCAAGGATGTGTTCCCTAGGAGGGTGGGTGTACCT
>mingled fasta file
GGGTGGTGTGTGCCACCCCTGATGAGACCGAAAGGTCGAAATGGGGTTTCGCCATGGGTCGGGACTTTAAATTCGGAGGATTCGTCCTTTAAACGTTCCT
CCAAGAGTCCCTTCCCCAAACCCTTACTTTGTAAGTGTGGTTCGGCGAATGTACCGTTTCGTCCTTTCGGACTCATCAGGGAAAGTACACACTTTCCGAC
GGTGGGTTCGTCGACACCTCTCCCCCTCCCAGGTACTATCCCCTTTCAAGGATGTGTTCCCTAGGAGGGTGGGTGTACCTCTTTTGGATTGCTCCGGCCT
TCCAGGAGAGATAGAGGACGACCTCTCCCCATACTGGAAACAACCTTGTGGTTCCTGTGGGTCACCCCTCCAAAGCAATAAAGGAAAAGAAAGGCGAAGA
Figure 1 shows the matching of the first three entries to demonstrate that the sequences of the output file corresponded to those of the input file. The boundaries of the individual files were reported on the “mingled.tsv” file:

Visualization of the mingling process. The figure shows the terminal's output of the shell commands used to display part of the content of the input file “viroid.fa”
The calculated lengths were compared with those deposited into the NCBI database:
To note the additional column “true length” present in “mingle_tested.tsv”. To assure that the reported boundaries corresponded to the actual location of the individual sequences, three random sequences were selected (corresponding to the GenBank IDs NC_22893.1, NC_003613.1, and NC_001351.1), and their reference fasta files were retrieved from the NCBI database and saved to fasta files. These were individually aligned to “mingled.fa” with:
Figure 2 shows that the boundaries for these sequences were, respectively, 2166-2577 (NC_22893.1), 8255-8624 (NC_003613.1), and 11979-12280 (NC_001351.1). These values were compared with those provided by mingle:

Boundaries of selected sequences. Three sequences were randomly selected from the bulk of the 47 viroid genomes available and were individually aligned to the “mingled.fa” file that represented the reference genome. The figure shows only the 5′ and 3′ ends of the sequences for cherry leaf scorch small circular viroid-like RNA
2166 2577 412 >NC_022893.1 Cherry leaf scorch small circular viroid-like RNA 1 isolate es.10, complete sequence
8255 8624 370 >NC_003613.1 Iresine viroid 1, complete genome
11979 12280 302 >NC_001351.1 Hop stunt viroid, complete genome
Mingle was also tested for its capability of providing different line width:
>Standard width of 80
GGGTGGTGTGTGCCACCCCTGATGAGACCGAAAGGTCGAAATGGGGTTTCGCCATGGGTCGGGACTTTAAATTCGGAGGA
TTCGTCCTTTAAACGTTCCTCCAAGAGTCCCTTCCCCAAACCCTTACTTTGTAAGTGTGGTTCGGCGAATGTACCGTTTC
>Custom width of 50
GGGTGGTGTGTGCCACCCCTGATGAGACCGAAAGGTCGAAATGGGGTTTC
GCCATGGGTCGGGACTTTAAATTCGGAGGATTCGTCCTTTAAACGTTCCT
>Custom width of 110
GGGTGGTGTGTGCCACCCCTGATGAGACCGAAAGGTCGAAATGGGGTTTCGCCATGGGTCGGGACTTTAAATTCGGAGGATTCGTCCTTTAAACGTTCCTCCAAGAGTCC
CTTCCCCAAACCCTTACTTTGTAAGTGTGGTTCGGCGAATGTACCGTTTCGTCCTTTCGGACTCATCAGGGAAAGTACACACTTTCCGACGGTGGGTTCGTCGACACCTC
The number of characters was the same in each of these files but, as expected, the number of lines was proportional to the line width:
15803/159
15803/317
15803/198
15803/144
The data provided so far showed that mingle could convert the individual genomes in a trustful way. The final outcome of this manipulation can be seen in Figure 3. It is customary to visualize mapped MPS reads using programs such as IGV; this step requires a reference genome. By providing a concatenated file with individual genomes, IGV reports each of the entries as a separate chromosome making the navigation cumbersome. Conversely, mingle provides a single cumulative chromosome that the operators can navigate with much ease. The following data will demonstrate other features of mingle not directly associated with the mapping of reads.
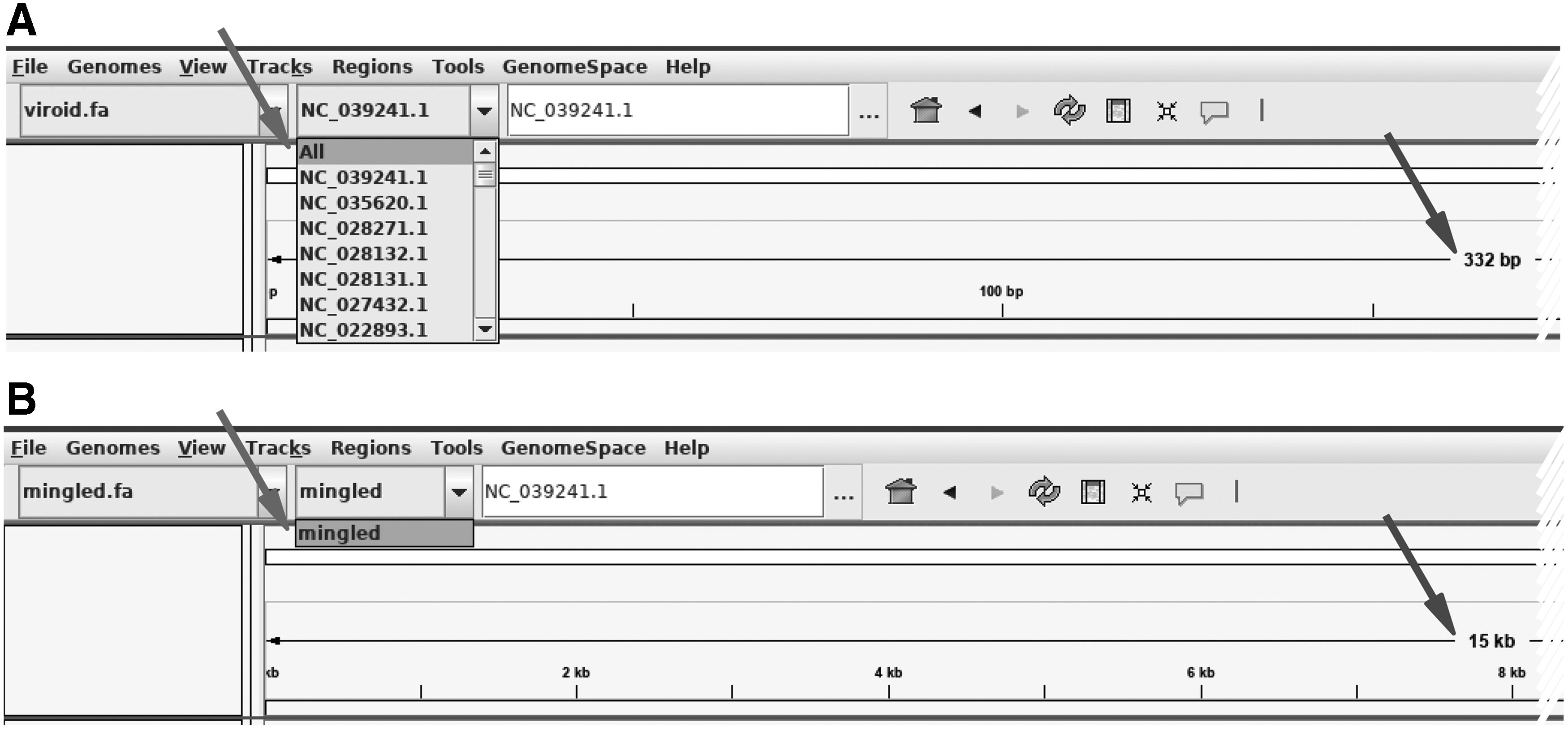
Differences in the visualization of the MPS reads according to the selected reference genome. This figure shows screenshots of integrative genome viewer with different reference genomes loaded in the environment. The file “viroid.fa” contained 47 individual viroid sequences
Identifying genomes parsing mingle's output was faster than enquiring remote servers: by running a Bash file purposely written to parse the local “mingled.tsv” file and to access the GenBank database, we found that the former approach could identify the IDs of selected genomes in a fraction of a second, whereas the remote access took an average of 21 ± 11 minutes (Supplementary File S3).
Since a fasta file can accommodate any custom sequence, the line width might not be constant; therefore, a direct stacking of the sequences could result in a visually unpleasant file. Mingle, in contrast, was capable of producing an output with a constant line width:
>First sequence
TGTGGGTCACCCCCCCAGCGAGTAATAAAAAGACAAGGGGTCGAGGACTCACC
GAGACGAGCGA
>Second sequence
GGCCGCTGGGCACTCCCCACCGTCCTTTTTTGCCAGTTCGCTCCAGGTTTCCCCGGGGATCCCTGAAGCGCTCCTCCGA
GCTTACCTGGCTTCCC
CGCTGAAACAGGGTTTTCACCCTTCCTTTCTTCGGGTTTCCT
>Third sequence
TCTCCCCCGCCGCTCTGGTAAGGCCCCTGCAGCGA
AGATCCCGCTAGTCGAGCGGACTTGTCGATCAACCAACGCGCTTTTCTTTTATTC
>Heterogeneous line width
TGTGGGTCACCCCCCCAGCGAGTAATAAAAAGACAAGGGGTCGAGGACTCACC
GAGACGAGCGA
GGCCGCTGGGCACTCCCCACCGTCCTTTTTTGCCAGTTCGCTCCAGGTTTCCCCGGGGATCCCTGAAGCGCTCCTCCGA
GCTTACCTGGCTTCCC
CGCTGAAACAGGGTTTTCACCCTTCCTTTCTTCGGGTTTCCT
TCTCCCCCGCCGCTCTGGTAAGGCCCCTGCAGCGA
AGATCCCGCTAGTCGAGCGGACTTGTCGATCAACCAACGCGCTTTTCTTTTATTC
CCTCTTGCGCGGCGGGGCGAGAGTGTTTTGCCTAAGGACCGCTGTTTGGGTCCTTGGGTGAAC
AACCTTGTGGTTCCTGTGGGTCACCCCGCCCCACGGA
>Homogeneous line width
TGTGGGTCACCCCCCCAGCGAGTAATAAAAAGACAAGGGGTCGAGGACTCACCGAGACGAGCGAGGCCGCTGGGCACTCCCCACCGTCCTTTTTTGCCAG
TTCGCTCCAGGTTTCCCCGGGGATCCCTGAAGCGCTCCTCCGAGCTTACCTGGCTTCCCCGCTGAAACAGGGTTTTCACCCTTCCTTTCTTCGGGTTTCC
TTCTCCCCCGCCGCTCTGGTAAGGCCCCTGCAGCGAAGATCCCGCTAGTCGAGCGGACTTGTCGATCAACCAACGCGCTTTTCTTTTATTCCCTCTTGCG
Since a fasta file could contain amino acid sequences, mingle was tested for its capability of dealing with them:
>transposase subunit 1
MSAKRIAMRKIREVLRLRLEAQLSFRQISICTKVSVGSIQKLLK
SAQALGLSWPLPTELDDGRLAALFYPQADTTISARHVVPDWPSVHQELKRKGVSKQLL
WEEYTQRYPNSCYSYSQFCDRYKSWCQLQKRSMRQIHKAGEKLFIDYCGPTVPIVSPT
TGEVRQAQVFVAVLGASNYTFAEATWSQSLPDWLQSHVRAFEFFGGTPALLIPDNLKS
GVSKACRYDPELNPSYQQLAEHYQVAVMPARPYKPKDKAKAEVGVQIVERWILARLRH
QTFFSLAELNQCIRALLNELNERPFKQLPGNRRDAFEQLDRPALGPLPVHPYRYVAIK
TVKVNIDYHVSYEQHHYSVPHQYVGQQLELHAGDTLLQVYHQQQLVASHPRKTIPGMS
TLPEHMPERHSKQQRWTPGRLKQWAADIGPGTLCWVSERLEEKAHVEQAYRLCLGLLS
LSREYPPSRVEACCQLANREGLVRLKQLKSVLASGRDQLPASPLSYPELPQEHENIRG
PQSFH
>transposase subunit 2
MTQQTLTRLRNLKLTGMADAVQQQLEQASTYEGLPFIERLSLLV
EHEQLSREQRKQTRLVKQARLKLQATVQELDYQSARNLERSQVANLAQGEWLRRGQNL
LITGPCGCGKTYLACALGYQACQQGHSTRYYRLPRLLLELSQAKVDGSYARLLGQLAR
IELLILDDWGLEAVNAEQRNALLEIMDDRYGKYSTIVVSQLPTEEWYASLGDNTLADA
ILDRLMHNAHRLSLKGESMRKRRSELTQSEQSS
>merged transposase
MSAKRIAMRKIREVLRLRLEAQLSFRQISICTKVSVGSIQKLLKSAQALGLSWPLPTELDDGRLAALFYPQADTTISARHVVPDWPSVHQELKRKGVSKQ
LLWEEYTQRYPNSCYSYSQFCDRYKSWCQLQKRSMRQIHKAGEKLFIDYCGPTVPIVSPTTGEVRQAQVFVAVLGASNYTFAEATWSQSLPDWLQSHVRA
FEFFGGTPALLIPDNLKSGVSKACRYDPELNPSYQQLAEHYQVAVMPARPYKPKDKAKAEVGVQIVERWILARLRHQTFFSLAELNQCIRALLNELNERP
FKQLPGNRRDAFEQLDRPALGPLPVHPYRYVAIKTVKVNIDYHVSYEQHHYSVPHQYVGQQLELHAGDTLLQVYHQQQLVASHPRKTIPGMSTLPEHMPE
RHSKQQRWTPGRLKQWAADIGPGTLCWVSERLEEKAHVEQAYRLCLGLLSLSREYPPSRVEACCQLANREGLVRLKQLKSVLASGRDQLPASPLSYPELP
QEHENIRGPQSFHMTQQTLTRLRNLKLTGMADAVQQQLEQASTYEGLPFIERLSLLVEHEQLSREQRKQTRLVKQARLKLQATVQELDYQSARNLERSQV
ANLAQGEWLRRGQNLLITGPCGCGKTYLACALGYQACQQGHSTRYYRLPRLLLELSQAKVDGSYARLLGQLARIELLILDDWGLEAVNAEQRNALLEIMD
DRYGKYSTIVVSQLPTEEWYASLGDNTLADAILDRLMHNAHRLSLKGESMRKRRSELTQSEQSS.
4. Discussion
This study introduced mingle, a utility for merging multi-fasta files while providing the boundaries of each original sequence. Mingle was designed to be a swift bioinformatic tool that can be used on computer clusters and as part of pipelines; it is relatively small (8.3 Kb), it does not require compilation, and works under the Unix/Linux environment, which is widely used in bioinformatic settings, but can be easily adapted to work also on Windows environment. The driving idea for developing mingle was the creation of custom genomes for MPS analysis; the user could patch together several chosen sequences and then use mingle to generate a single reference sequence while keeping track of the boundaries of each of the original genomes. When visualizing the obtained reads from the actual MPS experiment, this information could be used to determine the origin of the reads without the need for querying nucleotide databases, for instance by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi); enquiring a local file is much faster than connecting to a remote server and does not encounter connection problems.
Another important feature that drove the development of mingle was that its use should be made as easy as possible. Although expert bioinformaticians can approach the merging of sequences writing ad hoc procedures in a variety of ways, we wanted to provide a simple tool that could be adopted by users with general computer language literacy. With a simple syntax, mingle can be applied without requiring a deep bioinformatic knowledge and, therefore, it can be defined as a user-friendly application.
The data provided herein showed that mingle could reliably merge several sequences providing a list of boundaries and genome lengths that matched the value deposited in the NCBI database.
Footnotes
Acknowledgments
The authors thank the discussion forum “Biostars” (![]() ) for the support on some of the debugging issues encountered while writing mingle.
) for the support on some of the debugging issues encountered while writing mingle.
This study was supported by the Alfried Krupp von Bohlen und Halbach Foundation, Essen, the Deutsche Krebshilfe, Bonn (70112168), the Deutsche Forschungsgemeinschaft (DFG, grant number AL 465/9-1), the HEiKA Initiative (Karlsruhe Institute of Technology/University of Heidelberg collaborative effort), Dr Hella-Buehler-Foundation, Heidelberg, Ingrid zu Solms Foundation, Frankfurt, the DKFZ-MOST Cooperation, Heidelberg (grant number CA149), and the HIPO/POP-Initiative for Personalized Oncology, Heidelberg (H032 and H027).
Author Disclosure Statement
The authors declare there are no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.