
Abstract
Abstract
The purpose was to explore distinct molecular mechanisms of three lung cancer subtypes. GSE6044 microarray data downloaded from Gene Expression Omnibus (GEO) database were applied for identifying the differentially expressed genes (DEGs). Genetic global network was constructed to analyze the network annotation. The DEGs in the genetic global network related to small-cell lung carcinoma (SCLC), lung squamous cell carcinoma (SCC), and lung adenocarcinoma (AC) were screened. Protein–protein international networks of DEGs were constructed. Pathway enrichment analyses of DEGs in three subtypes were performed, followed by construction of interactional network among pathways. There were more DEGs screened in SCLC than in AC and SCC. The genetic global network with 341 genes and 1569 interaction edges was constructed. After annotating these DEGs into a protein interactional network, a total of 695 protein interactions related to these 36 DEGs were obtained. HSP90AA1 was the hub node with the highest degree of 81 in the annotation network. DEGs in SCLC and SCC were mainly enriched in some pathways, including cell cycle, DNA replication, and histidine metabolism; whereas DEGs in AC were enriched in complement and coagulation cascades, and extracellular matrix (ECM)-receptor interaction. Pathway interactional network was constructed with the hub node of a neuroactive ligand receptor interaction. The identified DEGs such as retinoid X receptor alpha (RXRA), cyclin-dependent kinase 2 (CDK2), histone deacetylase 2 (HDAC2), and KIT might be the target genes of lung cancer by participating in different pathways such as ECM-receptor interaction. Complement and coagulation cascades, and ECM-receptor interaction might be the specific pathways for AC; smoking might have a closer relationship with SCC.
1. Introduction
Lung cancer, also known as pulmonary carcinoma, is a malignant lung tumor that is always derived from epithelial cells and is difficult to be controlled. The most common symptoms of lung cancer are coughing up blood, shortness of breath, weight loss, and difficulty swallowing. The main causes of this disease included tobacco smoking, genetic factors, and exposure to asbestos, radon gas, and second-hand smoke. For therapeutic purposes, lung cancer could be classified into two broad classes: small-cell lung carcinoma (SCLC) and nonsmall-cell lung carcinoma (NSCLC). In addition, adenocarcinoma and squamous-cell carcinoma were the important subtypes of NSCLC. Commonly, SCLC is always treated by radiotherapy and chemotherapy, whereas NSCLC is treated with surgery with a better response.
Similar to other cancers, lung cancer is initiated by inactivation of tumor suppressor genes and activation of oncogenes. Mutations in the RAS proto-oncogene have confirmed to be an important cause of lung adenocarcinomas (ACs) (Sudhir et al., 2011). In addition, BRAF could encode an RAS-regulated kinase that mediated the activation of the malignant transformation kinase pathway and cell growth in SCLC (Brose et al., 2002). Besides, EGFR, which regulates various biological processes including cell proliferation, apoptosis, tumor invasion, and angiogenesis, is always found in NSCLC (Johnson et al., 2005). The EGFR mutations were clustered near the ATP cleft of the tyrosine kinase domain, and the EGFR TK inhibitors such as erlotinib have become a standard treatment for NSCLC (Lynch et al., 2004; Paz-Ares et al., 2006). Moreover, KEAP1 was also identified to participate in the regulatory pathway, and it plays an important role in the protection of cells against xenobiotic and oxidative damage (Taguchi et al., 2011). KEAP1 is a substrate adaptor protein for binding substrate to E3-ubiqutin ligase that could prevent undesired downstream effects (Massrieh et al., 2006). Interestingly, KEAP1 knockout in lung cancer experiments was shown to prevent cell damage caused by tobacco smoke by enabling Nrf2 accumulation (Thu et al., 2011).
The molecular mechanism research provided a theoretical basis for lung cancer, and classification has important implications for lung cancer treatment and diagnosis. Therefore, in this study, gene expression profiles of GSE6044 were downloaded to explore distinct molecular mechanisms analysis of three lung cancer subtypes. In a previous study, the gene expression profile of GSE6044 was researched for various molecular mechanisms of lung cancer. The experiment by Rohrbeck et al. (2008) showed the different gene expression profiles with specific biological characteristics of three lung tumor subtypes. And on this basis, genes predictive of oral squamous-cell carcinoma were identified (Chen et al., 2008). In addition, some subtype-specific genes, including p16, DBC1, and G0S2, were confirmed to be aberrantly methylated in NSCLC and lung squamous cell carcinoma (SCC) (Kusakabe et al., 2010). What is more, Zhou et al. (2014) identified featured biomarkers such as SNCA, ICAM1, and KIT in different types of lung cancer by combining with miRNA microarray profile. In this article, we aimed at analyzing the differences and similarities of molecular mechanisms in three lung cancer subtypes based on genetic global network, network annotation diagram, and pathway interaction network, and we further provided a theoretical basis for subtype-specific treatments.
2. Materials and Methods
2.1. Microarray data
The gene expression profiles of GSE6044 were downloaded from NCBI Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo) public repository with the platform of GPL201 [HG-Focus] Affymetrix Human HG-Focus Target Array (Rohrbeck et al., 2008). A total of 34 samples were studied, including 9 SCLC samples, 10 AC samples, 10 SCC samples, and 5 normal lung tissue samples.
2.2. Data preprocessing and differentially expressed genes screening
The raw data were read by Affy package and processed by the robust multi-array analysis method; then, normalization was performed on the processed data with a universal background (Zhang et al., 2012). Among the processed expression profile data, the probe with various Entrez Geneid lines was deleted, and the mean value was taken when multiple probes corresponded to one Entrez Geneid line. Student's test (Gao et al., 2014) was applied to screen differentially expressed genes (DEGs), and Benjamini-Hochberg (BH) (Stadtfeld et al., 2010) procedure was used to correct p-value. p < 0.05 and BH-corrected p-value <0.05 was the threshold of DEGs, respectively.
2.3. Construction of network annotation diagram
A total of 14 specific type cancers were downloaded from Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (www.genome.jp/kegg/pathway.html) for extracting the effective genetic information of genes and pathways. Among these genes and pathways, genes and their interaction in pathways were defined as nodes and edges for the genetic global network. Then, the gene expression levels in the three lung cancer types were annotated to this network based on the Human Protein Reference Database (HPRD, www.hprd.org), and network annotation diagrams were further constructed. In addition, DEGs in the network annotation diagram were screened with |log (fold change)| >2 or <0.5 and DEGs with BH-corrected p-value <0.1 were also screened.
2.4. KEGG pathway enrichment analysis
Expression Analysis Systematic Explorer (EASE), having similar analytic capability to that of DAVID, is a customizable tool for discovering enriched biological themes within gene lists (Hosack et al., 2003). It was used for KEGG pathways enrichment analysis of screened DEGs with p < 0.05 and |log (fold change)| >2 or <0.5. The threshold value of EASE was 0.1. Then, the pathways with BH-corrected p < 0.05 were screened as significant pathways.
2.5. Construction of interactional network among pathways in different lung cancer subtypes
If two DEGs in different pathways had a protein–protein interaction, these pathways were regarding as having the relationship of a co-changed interaction. In addition, protein–protein interaction networks related to these DEGs were constructed based on the HPRD database. Then, the cumulative geometric probability distribution model was used to evaluate the interaction significance between two pathways. The interactions of two pathways with p < 0.01 were regarded as significantly interacted pathways and defined as differently co-changed pathways. After removing intersected genes in two pathways, the cumulative geometric probability distribution model was calculated as the following formula:
In this formula, N represents the protein interaction number of all screened DEGs; M and N represent the protein interaction numbers of DEGs in two pathways, respectively; and K represents the interaction numbers of two pathways. Then, the interactional network among pathways in different lung cancer subtypes was constructed. Finally, pathway interactional networks of three lung cancer subtypes were screened.
3. Results
3.1. Screening of DEGs in three lung cancer subtypes
A total of 1161 up- and 1243 downregulated DEGs with p < 0.05 were screened in SCLC samples. In addition, a total of 649 up- and 677 downregulated DEGs with p < 0.05 were screened in AC samples. Moreover, 759 up- and 909 downregulated DEGs with p < 0.05 were screened in SCC samples. When BH-corrected p-value <0.05 was chosen as the threshold, a total of 567 up- and 565 downregulated DEGs were screened in SCLC samples. A total of 64 up- and 188 downregulated DEGs were screened in AC samples. Besides, 84 up- and 227 downregulated DEGs were screened in SCC samples (Table 1).
Number of Differentially Expressed Genes Screened in Small-Cell Lung Carcinoma, Lung Squamous Carcinoma, and Lung Adenocarcinoma
AC, lung adenocarcinoma; BH, Benjamini-Hochberg; SCLC, small-cell lung carcinoma; SCC, lung squamous cell carcinoma.
3.2. Construction of network annotation diagram
The genetic global network with 341 genes and 1569 interaction edges was constructed. In addition, the network annotation diagram of three lung cancer subtypes is shown in Figure 1 (A. SCLC samples; B. AC samples; C. SCC samples). There were more DEGs screened in SCLC samples than the other two subtypes. Table 2 showed the number of DEGs of three lung cancer subtypes in the genetic global network. A total of 26, 14, and 21 DEGs with |log (fold change)| >2 or <0.5 were screened in SCLC, AC, and SCC samples, respectively. The BH-corrected p-value <0.05 was taken as the threshold; whereas a total of 50, 11, and 16 DEGs were screened in SCLC, AC, and SCC samples, respectively.

Annotation of DEGs in genetic global network
Number of Differentially Expressed Genes Screened in Genetic Global Network
In addition, when the threshold was a BH-corrected p-value <0.05 and |log (fold change)| >2 or <0.5, a total of 22, 6, and 8 DEGs were screened in SCLC, AC, and SCC samples, respectively. After annotating these DEGs into a protein interactional network based on the HPRD database, a total of 695 protein interactions related to these 36 DEGs were obtained. Figures 2–4 showed the annotation results of DEGs in three lung cancer subtypes. In this annotation network, HSP90AA1, retinoid X receptor alpha (RXRA), and BCL2 were the hub nodes with a higher degree of 81, 67, and 64, respectively. Moreover, Table 3 showed the differently expressed levels of 20 DEGs with a higher degree in three lung cancer subtypes. In these lung cancer subtypes, a total of 10 DEGs, such as RXRA, cyclin-dependent kinase 2 (CDK2), HDAC2, and CDK4 had the same direction of disorder. Moreover, only four DEGs, including FGFR3, EPAS1, COL4A1, and BIRC5, with |log (fold change)| >2 were screened.

Protein–protein interactional network of DEGs for SCC. The red and green node represent the up- and downregulated DEGs in genetic global network, respectively.

Protein–protein interactional network of DEGs for AC. The red and green node represent the up- and downregulated DEGs in genetic global network, respectively.
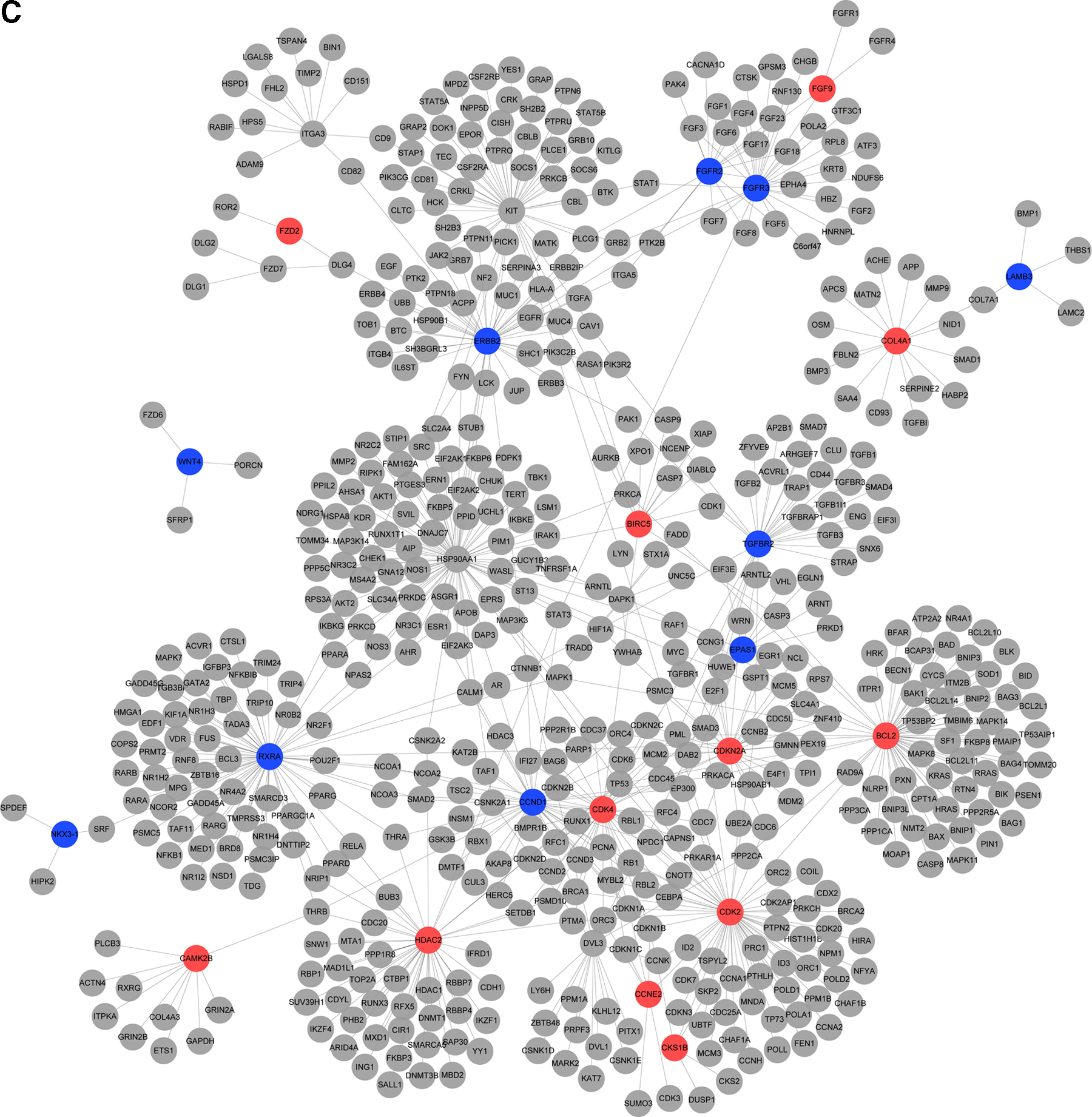
Protein–protein interactional network of DEGs for SCLC. The red and green node represent the up- and downregulated DEGs in genetic global network, respectively.
Top 20 Differentially Expressed Genes Related to Small-Cell Lung Carcinoma, Lung Squamous Cell Carcinoma, and Lung Adenocarcinoma in Protein–Protein Interactional Network
BH_P, BH-corrected p value; dir, direction; fc, fold change.
3.3. KEGG pathway enrichment analysis
Table 4 showed the KEGG pathways enriched by DEGs in three lung cancer subtypes. The results showed that DEGs in SCLC were mainly enriched in some pathways, including cell cycle, DNA replication, and histidine metabolism. In addition, KEGG pathway enrichment results of SCC present similar characteristics with SCLC. However, DEGs in AC were mainly enriched in two pathways, including complement and coagulation cascades, and extracellular matrix (ECM)-receptor interaction.
Kyoto Encyclopedia of Genes and Genomes Enrichment by Differentially Expressed Genes in Small-Cell Lung Carcinoma, Lung Squamous Cell Carcinoma, and Lung Adenocarcinoma
ECM, extracellular matrix.
3.4. Construction of interactional network among pathways in different lung cancer subtypes
Interactional networks among pathways in different lung cancer subtypes are shown in Figure 5–7. In the interactional network of SCLC, neuroactive ligand receptor interaction, citrate cycle, pyruvate metabolism, arachidonic acid metabolism, and vaso pressin regulated water reabsorption were the hub nodes with higher degrees of 22, 9, 8, 6, and 5, respectively. Moreover, in the interactional network of AC and SCC, the pathway of neuroactive ligand receptor interaction was also the hub node with the highest degree of 29 and 19.

Pathway interactional network for SCLC. The nodes represent the enriched pathways, and the edges represent the relationship with the co-changed pathways.

Pathway interactional network for AC. The nodes represent the enriched pathways, and the edges represent the relationship with the co-changed pathways.
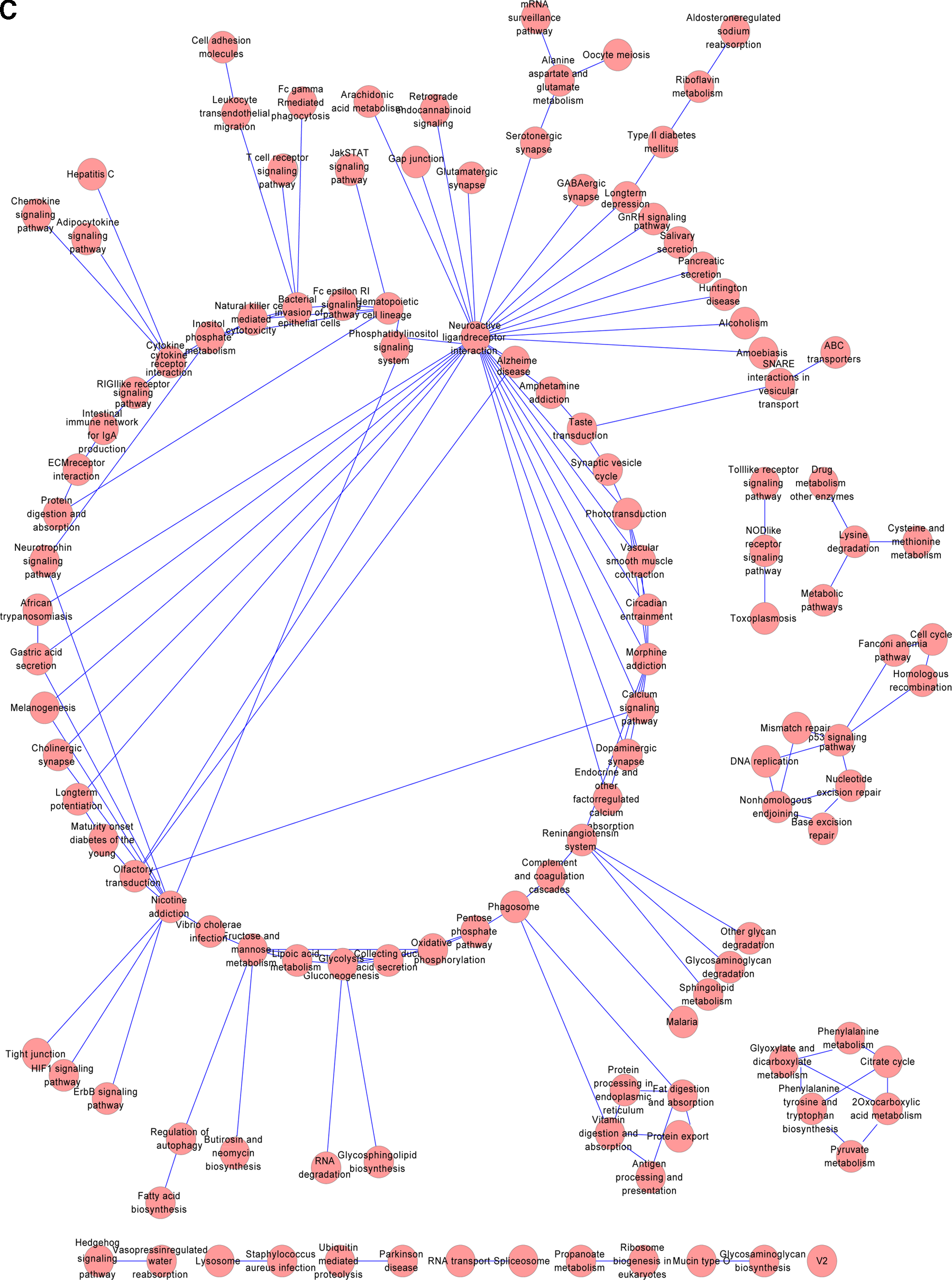
Pathway interactional network for SCC. The nodes represent the enriched pathways, and the edges represent the relationship with the co-changed pathways.
4. Discussion
Lung cancer is an enormous threat to human health and it is associated with a high recurrence rate (Lu et al., 2012). Therefore, there is an urgent need to research the molecular mechanisms of different lung cancer subtypes in the level of gene, pathway, and network for specific treatment. In this artcile, DEGs between normal samples and different subtypes of lung cancer samples were identified. Further, we found that the expression of RXRA, CDK2, and histone deacetylase 2 (HDAC2) showed no difference; whereas BCL2 and KIT were differently expressed in the three lung cancer subtypes according to the constructed network annotation diagram.
RXRA, also known as NR2B1, served as a common heterodimeric partner for a number of nuclear receptors (Evans and Mangelsdorf, 2014). In addition, RXRA heterodimers act as ligand-dependent transcriptional regulators and increase the DNA-binding efficiency of its partner (Lefebvre et al., 2010). Dmitrovsky (2013) also showed that RXRA heterodimer could trigger G1 cyclin destabilization by activating with a rexinoid. In this study, all the expression levels of RXRA in three lung cancer subtypes were down, further regulated cell cycle, and participated in the development of lung cancer.
Besides, CDK2 and HDAC2 also showed the same direction in three subtypes of lung cancer and participated in the DNA replication pathway. CDK2 encodes a member of a family of serine/threonine protein kinases that takes part in cell cycle regulation, especially in meiosis (Goldsmith et al., 2007). The activation of the CDK family could control the timing of entry into mitosis/meiosis by regulating cyclin E or A (Cánepa et al., 2007). In addition, NPM1 phosphorylation mediated by cyclin E/CDK2 improved dissociation from unduplicated centrosomes, and it further induced centrosome duplication (Frehlick et al., 2007). Besides, HDAC2, whose product belongs to the histone deacetylase family, acts by forming large complexes, and it further promotes the deacetylation of lysine residuces of core histones (Ito et al., 2000). By associating with SIN3, MAD, YY1, and N-COR, HDAC2 products could form transcriptional repressor complexes, and the formed complex could interact with DNMT1 in DNA replication and regulate transcriptional repressor activity (Turek-Plewa and Jagodzinski, 2005). Thereby, RXRA, CDK2, and HDAC2 might participate in pathways of cell cycle and DNA replication, and they could further regulate three lung cancer subtypes in the same manner.
In SCLC, DEGs, including KIT and BCL2, were significantly upregulated; whereas these DEGs in AC and SCC were downregulated. KIT (v-kit hardy-zuckerman 4 feline sarcoma viral oncogene homolog) encodes the human homolog of the proto-oncogene c-kit (Curtin et al., 2006). KIT could activate downstream singling pathways such as PI3K-AKT, MAPK-ERK, and JAK-STAT by combining with SCF, and it could further improve cell growth and proliferation (Armstrong et al., 2006). Moreover, a cell experiment confirmed that Gleevec could inhibit the growth of SCLC by inhibiting the expression of KIT (Wang et al., 2005). Moreover, Bcl-2 (B-cell CLL/lymphoma 2) encodes some anti-apoptotic protein, and its family is always over-expressed in various cancer cells (Fuenzalida et al., 2007). Over-expressed Bcl-2 could improve the ability against apoptosis and increase the resistance of chemoradiotherapy (Jeong et al., 2010). Sartorius and Krammer (2002) showed that the increasing expression of Bcl-2 could increase various chemotherapies, including etoposide, adriamycin, and cis-platinum. Thereby, DEGs such as KIT and Bcl-2 might be the target genes in SCLC.
The GO enrichment results showed that GO terms enriched in SCLC and SCC mainly included cell cycle, DNA replication, and tyrosine metabolism. To prevent these pathways, treatment methods such as erlotinib (Zhou et al., 2011), anaplastic lymphoma kinase (Kwak et al., 2010), and scutellaria baicalensis (Gao et al., 2011) were researched for lung cancer treatment. However, DEGs in AC were enriched in pathways of complement and coagulation cascades, and ECM-receptor interaction. Complement and coagulation cascades played a critical role as a tie in the invasion and metastasis of cancer cells (Currie et al., 1990). What is more, the number of possible ECM-receptor interactions was potentiated by different ECM components competing for one receptor (Lochter and Bissell, 1995). In addition, this pathway also influenced cellular behavior by changing the organization of the cytoskeleton and activating the second messenger (Taddei et al., 2007). Thereby, pathways of complement and coagulation cascades, and ECM-receptor interaction might play an important role in pathogenesis of AC.
Interestingly, the pathway of neuroactive ligand-receptor interaction had a close interaction with other pathways in three lung cancer subtypes. A previous study showed that genes of the neuroactive ligand-receptor interaction pathway were the important components of the G-protein coupled receptors (Wei et al., 2012). The GPCRs could trans-activate the EGFR pathway and further lead to the improvement of cell growth and motility in lung cancer (Krysan et al., 2005). From the results of the pathway network of SCC, the enriched nicotine addiction and morphine addiction confirmed that smoking was a key factor in the development of SCC.
The identified DEGs such as RXRA, CDK2, HDAC2,BCL2, and KIT might be the target genes of lung cancer by participating in different pathways such as the ECM-receptor interaction. Complement and coagulation cascades, and ECM-receptor interaction might be the specific pathways for AC. And smoking might have a closer relationship with SCC. However, these results need to be further confirmed by experimental studies.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Footnotes
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Authors' Contributions
L.W. conceived and designed research and drafted the article. Y.P. conceived and designed research and acquired data. S.L. and S.Z. acquired data, analyzed and interpreted data and statistical analysis. Y.Y. revised the article for important intellectual content. All authors read and approved the final article.