
Abstract
Abstract
We attempted to analyze the aberrant pathways and genes underlying the successive stages of colorectal cancer (CRC). The CRC related microarray data (GSE77953) were retrieved from Gene Expression Omnibus database, which included 17 colonic adenoma, 17 carcinoma, 11 CRC metastases, and 13 normal colonic epithelium samples. The differential expression patterns in colonic adenoma, carcinoma, and metastases were analyzed. Gene functional interaction (FI) and coexpressed network were constructed. Pathway enrichment analysis was performed to investigate the perturbed pathways, and disease-related genes were explored based on the Comparative Toxicogenomics Database. Total 438 genes were identified to be differentially expressed in colonic adenoma, 885 in carcinoma and 736 in metastases. The upregulated genes in adenoma were significantly related with ribosome, oxidative phosphorylation, and protein export related pathways. The downregulated genes in carcinoma and metastases were enriched in the same pathways, such as nitrogen metabolism, mineral absorption, and steroid hormone biosynthesis. FI network was constructed with 219 and 3914 edges, which were further divided to 12 modules. The genes in module 0 were closely related with ribosome, protein export, and RNA transport. Coexpressed genes were enriched in ribosome, protein export, and mineral absorption pathways. Total eight common upregulated genes were found to be the CRC-related genes such as RNF43 (ring finger protein 43), EIF3H (eukaryotic translation initiation factor 3 subunit H), and STRAP (serine/threonine kinase receptor associated protein). The common downregulated genes included ABCG2 (ATP binding cassette subfamily G member 2), GCG (glucagon), and SULT1A1 (sulfotransferase family 1A member 1). Oxidative phosphorylation, nitrogen metabolism, mineral absorption, and protein synthesis may significantly be perturbed in the progression of CRC. The overexpression of EIF3H may be the predictor for CRC formation.
1. Introduction
Colorectal cancer (CRC) is a leading cause for cancer related death. CRC commonly develops from a benign tumor and then progresses to adenoma, carcinomas, and metastatic CRC over time (Mcguire, 2016). It is estimated that about 136,830 individuals will be diagnosed with CRC, and 50,310 cases die of this disease in 2014 (Siegel et al., 2014). Although there is a decline in the death rate by ∼2% per year, no approved treatment options are available for patients with metastatic CRC (Grothey et al., 2013). Thus, understanding CRC progression from normal colonic epithelium may be helpful in the prevention of CRC transformation.
Genetic alterations such as gene mutations and chromosomal instability drive the development and transformation of CRC. Protein kinase C (PK-C) plays a key role in the growth and differentiation of normal epithelial cells, and the altered activity of PK-C contributes to the transformation of colonic adenomas and carcinomas (Kopp et al., 1991). The loss function of the base-excision repair gene (NTHL 1) increases the trend of transition to adenomas and CRC (Weren et al., 2015). The overexpression of Kitenin and ErbB4 CYT-2 at mRNA level has been proposed to be the marker for predicting the transition from colonic adenomas to CRC (Bae et al., 2016). However, systematic genetic changes in the progression of CRC have not been fully clarified.
Thus, in the present study, we attempted to give an integrative view to understand the advanced CRC progression from normal colonic epithelial tissues. Compared with normal tissues, the differentially expressed genes (DEGs) in adenoma, carcinoma, and CRC metastases were identified, respectively. The coexpression and functional interaction (FI) networks were constructed, and the CRC-related genes were mined. We expect that our findings can pave the novel way to understand the progression of CRC and help discover the novel target for preventing CRC transition.
2. Methods
2.1. Microarray data and data processing
The CRC related gene expression data were obtained from National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo) with the accession number of GSE77953 (Qu et al., 2016). The microarray data were produced from 58 tissue samples (17 Adenoma, 17 Carcinoma, 11 CRC metastases, and 13 normal colonic epithelium) that cover the major stages of CRC progression.
For the GSE77953 dataset, the raw CEL format data were downloaded based on the platform of Affymetrix Human Genome U133A Array. The raw dataset preprocessing was performed using the function of affy package in R (Gautier et al., 2004), including background correction, normalization, and expression estimates.
2.2. DEG analysis
The differential expression patterns induced by CRC successive stages were measured by comparing gene expression dataset of adenoma, carcinoma, and CRC metastases with normal tissues. The DEGs were identified by t test implemented in limma package (Smyth, 2005). Benjamini and Hochberg (BH) procedure was applied to control the statistical significance. The DEGs were identified with adjusted p value <0.05 and log2|fold change| ≥ 1.
2.3. Venn diagram analysis
VennPlex is an available open source for Venn diagram generation (www.irp.nia.nih.gov/bioinformatics/vennplex.html) (Cai et al., 2013). To identify the common and uniquely altered genes between different stages of CRC, the Venn diagram was constructed by VennPlex software. The number of upregulated, downregulated, and contraregulated genes in intersections was provided after Venn diagram analysis.
2.4. Pathway enrichment analysis
Database for Annotation, Visualization and Integrated Discovery (DAVID) is a web-accessible program that provides functional annotation for a large set of genes (Da Wei Huang and Lempicki, 2008). The DAVID online tool (version: 6.8) was used for analyzing the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways involved with DEGs. Pathways with count ≥2 and p value <0.05 were considered to be significant.
2.5. Gene FI network
ReactomeFI (Wu et al., 2014), as a Cytoscape app, is used for functional network analysis. The gene expression data of all the DEGs were collected, and the gene FI network was constructed by ReactomeFI app. FI network modules were generated by the application of Monte Carlo Localization graph clustering algorithm based on module size and average correlation. Subsequently, KEGG pathway enrichment was calculated for function modules.
2.6. Coexpressed gene analysis
Pearson correlation coefficients (PCC) were calculated to indicate the correlations between DEGs. As the previous study described (Liang et al., 2015), the p values were calculated based on Z-scores. The coexpressed gene pairs were selected when |PCC| > 0.9 and p < 0.05.
2.7. Disease-related gene analysis
Comparative Toxicogenomics Database (CTD) collects the chemical–gene interactions and gene–disease interactions (Davis et al., 2011). To explore the genes closely related with CRC occurrence, the DEGs closely related with the process of CRC successive stages were screened out based on the information provided by CTD.
3. Findings
3.1. Identification of DEGs
Compared with normal controls, 438 DEGs were found in Adenoma, 885 in Carcinoma, and 736 in CRC metastases. Venn diagram illustrates that total 312 genes are upregulated and 40 are downregulated in various stages of CRC (Fig. 1).
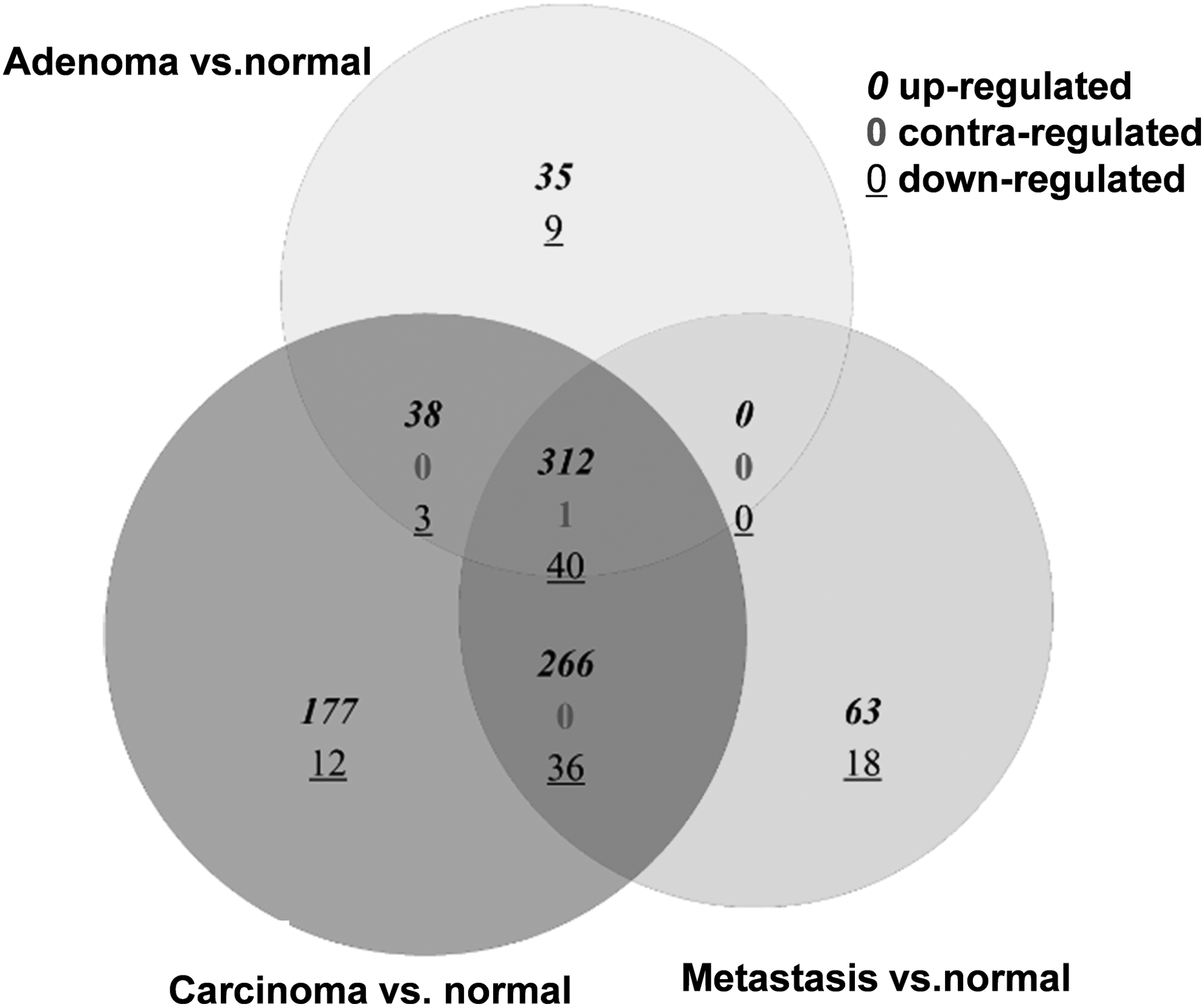
Venn diagram of the differentially expressed genes in various stages of colorectal cancer.
3.2. Significant pathways enriched by DEGs
The KEGG pathways that were significantly enriched by DEGs in Adenoma, Carcinoma, and CRC metastases were analyzed, respectively. As shown in Figure 2, the upregulated genes in Adenoma were mainly related with RNA transport, Ribosome, and protein export, which were similar with a part of pathways enriched by the upregulated genes in CRC metastases. Other pathways enriched by overexpressed genes in CRC metastases included the disease-related pathways, cell cycle, and proteasome that were also enriched by the upregulated genes in Carcinoma. Pathways enriched by downregulated genes in Carcinoma and CRC metastases were the same such as Nitrogen metabolism, Mineral absorption, Steroid hormone biosynthesis, Chemical carcinogenesis, and Aldosterone-regulated sodium reabsorption (Fig. 2).

The significant pathways enriched by the up- and downregulated genes in colonic adenoma, carcinoma, and metastases. Grey: significantly enriched pathway; black: no pathway significantly enriched.
In addition, the common upregulated genes in various stages of CRC were closely related with protein synthesis related pathways such as hsa03010: Ribosome, hsa03013: RNA transport, hsa03060: Protein export, and hsa00190: Oxidative phosphorylation. The common downregulated genes were enriched in two pathways, such as hsa04976: Bile secretion and hsa04972: Pancreatic secretion.
3.3. FI network and module analysis
FI network was produced, which was composed of 3914 edges interacting with 219 nodes (Fig. 3). The FI network contained 12 connected modules, which varied in size from 7 to 108 regional nodes (Table 1). Figure 4 exhibits the significant pathways for 12 modules. Module 0 was the most significant module with average correlation of 0.811, which contained 108 genes that were closely related with protein synthesis related pathways.

The gene functional interaction network. Oval nodes, differentially expressed genes; upper triangle, upregulated genes; lower triangle, downregulated genes.
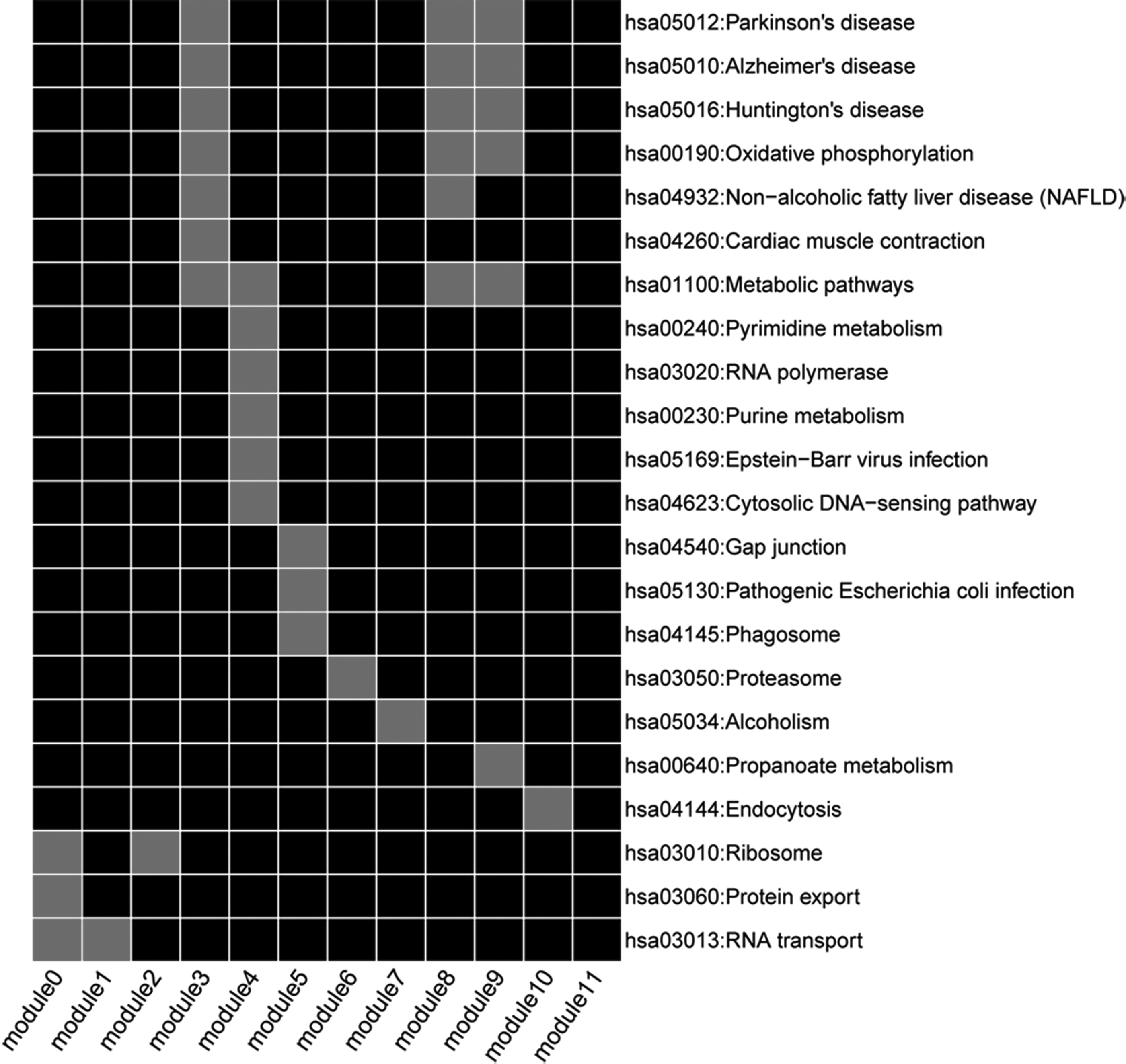
The significant pathways for different module genes of functional interaction network. Grey: significantly enriched pathway; black: no pathway enriched.
Module Analysis of the Functional Interaction Network
3.4. Gene coexpressed network
As shown in Figure 5, the gene coexpressed network was constructed with 270 nodes and 1549 interaction pairs. One hundred and eighty-nine of 270 nodes were the common DEGs in various stages of CRC, and these genes were significantly enriched in hsa03010: Ribosome, hsa03060: Protein export, and hsa04978: Mineral absorption.

The coexpression gene network for differentially expressed genes. Upper triangle, common upregulated genes; lower triangle, common downregulated genes; oval nodes, differentially expressed genes.
3.5. CRC related genes
The CRC related genes were mined in adenoma, carcinoma, and metastases stages of CRC, respectively. As shown in Table 2, 18 genes were documented to be related with adenoma, 25 with carcinoma, and 21 with CRC metastases. Total eight genes were all upregulated in the adenoma, carcinoma, and metastases stage of CRC, such as GGH (gamma-glutamyl hydrolase), RRM2 (ribonucleotide reductase regulatory subunit M2), RNF43 (ring finger protein 43), CDKN1B (cyclin dependent kinase inhibitor 1B), EIF3H (eukaryotic translation initiation factor 3 subunit H), STRAP (serine/threonine kinase receptor associated protein), ODC1 (ornithine decarboxylase, structural 1), and PCNA (proliferating cell nuclear antigen). The common downregulated genes related with various stages of CRC contained ABCG2 (ATP binding cassette subfamily G member 2), GCG (glucagon), and SULT1A1 (sulfotransferase family 1A member 1).
Disease-Related Genes in Different Stages of Colorectal Cancer
4. Discussion
Currently, the studies about systematic analysis of the gene expression alteration during the successive stages of CRC are rare. In this article, we performed an integrated genomic analysis of CRC progression by bioinformatic methods. Our data showed that total 438 genes were identified to be differentially expressed in colonic adenoma, 885 DEGs were found in carcinoma, and 736 in CRC metastases. The numbers of DEGs in carcinoma and CRC metastases were higher than that in colonic adenoma. We speculated that the pathological mechanism of CRC became complex over time. Pathway enrichment analysis showed that only 5 pathways were enriched by DEGs in adenoma, and there were 23 and 16 pathways dysregulated in carcinoma and CRC metastases, respectively, which was in accordance with our hypothesis.
In our article, oxidative phosphorylation was a pathway that was significantly enriched by upregulated genes in various stages of CRC. It is reported that the oxidative phosphorylation was altered in the process of cancerous growth (Polyak et al., 1998). Recent evidence has shown that mitochondrial oxidative phosphorylation is responsible for the energy and proliferation of various tumor cells such as acute myeloid leukemia, lymphoma, breast, melanoma, and pancreatic ductal adenocarcinoma (Molina et al., 2016). IACS-010759 has been suggested to be a novel candidate for acute myeloid leukemia by targeting oxidative phosphorylation (Molina et al., 2015). The alterations of oxidative phosphorylation may show the implication of abnormal metabolic and apoptotic process in cancer cells. Pathway analysis showed that the metabolism processes, such as nitrogen metabolism and mineral absorption, were dysregulated in both colonic carcinoma and metastases involved with the downregulated genes.
The basic body nutrients include nitrogen and minerals, which are changed in cancer cells and influence carcinogenesis. Nitrogen metabolism plays a key role in maintaining a constant body protein mass of healthy individuals (Miyazaki and Esser, 2008). It is reported that 30% of all the cancer patients are present with negative nitrogen balance (Blackburn et al., 1977). A recent study has indicated that hypoxia-inducible factors (HIF) regulate nitrogen metabolism in endothelial cells that affect the metastatic success of tumor cells (Shay and Simon, 2012). Although the precise role of mineral has not been determined, mineral metabolism may play a key role in tumor progression. It is reported that mineral deficiencies cause the DNA damage and increase the risk for cancer development (Ames and Wakimoto, 2002). Colonic carcinoma and metastases may share the similar mechanism of CRC progression.
The FI and gene coexpression network also showed that hsa00190: Oxidative phosphorylation and hsa04978: Mineral absorption were the significant pathways, suggesting that these pathways may play key roles in CRC development. In addition, the function interaction genes and coexpression genes were significantly enriched by protein synthesis related pathways such as hsa03010: Ribosome and hsa03060: Protein export. One important mechanism of the cancer formation is the dysregulation of translation (Van et al., 2010). The protein synthesis rate is increased by mediating the expression of ribosomal protein and translation factors. It is reported that the expression of ribosomal protein genes is increased in CRC (K et al., 1991). The protein synthesis initiation factor eIF-4E is found to be overexpressed in the early stage of CRC development (Rosenwald et al., 1999). The expression of eIF-4E is upregulated in colon adenomas and carcinomas. Thus, protein synthesis is an event in the early stage of colon carcinogenesis.
EIF3H was a significant node and showed multiple interactions with other genes in FI network. Pathway analysis showed that EIF3H was enriched in hsa03013: RNA transport pathway, which was related with protein synthesis. EIF3H functions in regulating cell growth and viability and its overexpression have been found to be a feature in breast, prostate, and liver cancer (Hiroyuki et al., 2003; Savinainen et al., 2004). Our work suggested that EIF3H was a disease related gene, which was determined by previous reports. Yao et al. have found that the genetic variety of EIF3H is linked to the CRC formation (Yao et al., 2014). EIF3H is a plausible causative gene, and the abnormal expression of EIF3H is closely related with the risk for CRC (Pittman et al., 2010). In our article, EIF3H was a common upregulated gene in the various stages of CRC. The overexpression of EIF3H may be the predictor for CRC progression.
5. Conclusion
In conclusion, oxidative phosphorylation may be a significant pathway in the process of CRC development and progression. Nitrogen metabolism and mineral absorption may be dysregulated in the process from colonic carcinoma to metastases. Protein synthesis is an early event in colon carcinogenesis, and the overexpression of the related EIF3H may be the predictor for CRC formation.
Footnotes
Author Disclosure Statement
The authors declare there are no competing financial interests.