
Abstract
This study aimed at investigating the crucial mechanisms underlying non-small cell lung cancer (NSCLC). NSCLC-related microarray data GSE27262 were downloaded from Gene Expression Omnibus, including 7 NSCLC 1a samples, 18 NSCLC 1b samples, and their matched normal samples. The common differentially expressed genes (DEGs) between NSCLC 1a and NSCLC 1b samples were identified, followed by protein-protein interaction (PPI) network construction, functional enrichment analysis, and weighted gene co-expression network analysis (WGCNA). Further, the key DEGs were confirmed based on the lung adenocarcinoma (LUAD) data from the Cancer Genome Atlas (TCGA) database, followed by clinical prognostic analysis. There were 802 (NSCLC 1a) and 734 (NSCLC 1b) DEGs identified. By intersection analysis, we obtained 255 upregulated and 97 downregulated common DEGs. Upregulated DEGs were significantly enriched in the plasma membrane and extracellular region, whereas the downregulated DEGs were significantly enriched in the cytoskeleton and cell cycle process. Topoisomerase (DNA) II alpha (TOP2A) and cyclin B1 (CCNB1) were hub nodes in the PPI network. Based on WGCNA, 5 modules were obtained. In the module MEgreen, DEGs were significantly enriched in cytokine-cytokine receptor interaction and focal adhesion. Notably, 1797 DEGs were identified based on the LUAD data from the TCGA database; among them, 285 DEGs were common DEGs identified from GSE27262 data. Upregulation of TOP2A and CCNB1 was correlated with poor survival of patients. The hub genes and key pathways identified in this study are helpful for a comprehensive knowledge of the molecular mechanisms of NSCLC.
1. Introduction
According to histological classification criteria, there is about 80.4% non-small cell lung cancer (NSCLC) of the total lung cancer, and ∼75% of the patients are found to be in advanced stage, with a low 5-year survival rate (Hisayuki et al., 2005). Surgical treatment is one of the therapeutic options for NSCLC (Danesi and Pasqualetti, 2009; Shih et al., 2012). However, with no specific symptoms associated with the early stage of NCLSC and the lack of accurate and convenient diagnostic methods for its early detection, a large number of patients are diagnosed in the late stage of NSCLC, and surgical resection is no longer feasible (Keith and Miller, 2013).
At present, NSCLC is generally believed to be related to smoking, environment, heredity, and other factors (Keith and Miller, 2013). With the development of cell biology and molecular biology, more and more research shows that the development of NSCLC may be interrelated to abnormal gene expression (Hisayuki et al., 2005; Gerber and Minna, 2010; Oxnard et al., 2013). With recent advances in molecular biology, molecular target therapy for NSCLC is equally significant. For example, approximately 30%–50% of Asian NSCLC patients and 10%–20% of North American and western European NSCLC patients are shown to have mutation of epidermal growth factor receptor (EGFR) gene (Oxnard et al., 2013). But, with the increasing use of EGFR tyrosine kinase inhibitors, such as erlotinib and gefitinib, these patients are found to respond well (with a response rate of 10%, and an estimated 2-month prolongation in median survival over placebo) (Gerber and Minna, 2010; Janku et al., 2010). It is also found that 3%–7% of NSCLC patients have an activated anaplastic lymphoma kinase (ALK) gene; however, treatment with crizotinib (an ALK inhibitor) elevates their response rate from 10% to 55% (compared with conventional chemotherapy) and increases their 6-month progression-free survival rate to 72% (Gerber and Minna, 2010). Although significant success has been achieved in targeted therapy for NSCLC, the exact molecular mechanisms are still far from being fully understood. Therefore, it is essential to find more potential targets to develop more effective treatment strategies.
In previous studies, Wei et al. (2012, 2014) developed microarray data GSE27262 to show that methylosome protein 50 may transform cells in an independent manner, and protein arginine methyltransferase 5 was a potential oncoprotein that could increase the cascade of G1 cyclin/cyclin-dependent kinase and inositol phosphate 3 kinase/AKT signal. However, they did not look at the differentially expressed genes (DEGs) of NSCLC specifically. In this study, for the sake of a better comprehension of the DEGs influence on pathogenesis of NSCLC, we downloaded these microarray data from Gene Expression Omnibus (GEO) and identified the DEGs between the NSCLC patients and matched normal samples. Subsequently, we performed functional enrichment analysis, protein-protein interaction (PPI) network analysis, and weighted correlation network analysis (weighted gene co-expression network analysis [WGCNA]). The hub genes and key pathways identified in this study are helpful for a comprehensive understanding of the molecular mechanisms of NSCLC.
2. Materials and Methods
2.1. Affymetrix microarray data
Microarray data GSE27262 (Wei et al., 2012, 2014) were downloaded from the public GEO database (www.ncbi.nlm.nih.gov/geo/), which was generated from 7 paired NSCLC 1a cancer samples and matched normal samples, and 18 paired NSCLC 1b cancer samples and matched normal samples All samples were tested on the platform of Affymetrix Human Genome U133 Plus 2.0 Array (GPL570 [HG-U133_Plus_2]).
2.2. Data pre-processing and differential expression analysis
Data pre-processing was performed by using the robust multiarray average algorithm in R package (Hobbs et al., 2003). The DEGs in NSCLC 1a and 1b stage samples were, respectively, identified compared with their matched normal samples by using the paired t-test according to the limma package (Hobbs et al., 2003). Finally, the DEGs with |log2-fold-change (FC)| > 2 and p < 0.05 were considered significant. Subsequently, the common DEGs were obtained from NSCLC 1a and NSCLC 1b stage samples.
2.3. Functional enrichment analysis for common DEGs
We used a database for annotation, visualization, and integrated discovery (DAVID, version 6.8) (Huang et al., 2008; Wei et al., 2009) to investigate the enriched gene ontology (GO) that included molecular function, cellular component, and biological processes terms (Csermely et al., 2005). Then, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (Tanya et al., 2005) was also enriched by common DEGs. Count ≥2 and p < 0.05 were used as the cut-off values.
2.4. Construction of PPI networks
The interactive relationships of common DEGs were explored by a search tool for the retrieval of interacting genes (STRING, version 10.0). The DEGs interactions with a combined score >0.4 were considered statistically significant. Then, the PPI network was built by Cytoscape software (version 3.2.0). If the nodes have a higher PPI score, the more likely they are the key hub genes. We used MCODE Bader and Hogue (2003) in Cytoscape to analyze the subnetwork module in the PPI network.
2.5. WGCNA network construction
The WGCNA package of R (7) was used to construct the co-expression network with DEGs. Limma packet was used to obtain the significant DEGs with the cut-off values of |log (FC)| > 1 and p < 0.05. Then, the adjacency between each gene was calculated, and the different module was constructed and selected with |coefficient| > 0.7 and p < 0.05. Finally, the KEGG pathway was also enriched by the genes in the selected modules (p < 0.05) (Langfelder and Horvath, 2008).
2.6. Confirmation of the identified DEGs by other datasets
To confirm the differential expression of key DEGs, the lung adenocarcinoma (LUAD) data from the Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) included 518 LUAD tissues and 58 adjacent normal tissues. The DEGs between LUAD tissues and 58 adjacent normal tissues were also identified by the paired t-test in the limma package, with the cut-off values of |log2 FC| > 2 and p < 0.05. The intersection analysis for the DEGs identified from LUAD data and the common DEGs identified from GSE27262 data was conducted. Further, based on the expression data and clinical information of LUAD data, patients were divided into high expression group and low expression group according to the mean expression value of each gene. The association between differential expression of key DEGs (such as TOP2A and CCNB1) and prognosis of NSCLC patients were then performed by survival analysis using Survival package (version 2.36-5, https://cran.r-project.org/web/packages/survival/index.html) (Therneau and Lumley, 2011). The survival curve was conducted by using the Kaplan-Meier method, and p < 0.05 was considered statistically significant.
3. Results
3.1. Identification of DEGs from microarray data
A total of 802 DEGs in NSCLC 1a group and 734 DEGs in NSCLC 1b group were obtained, and the heat map revealed that DEGs in the NSCLC samples and normal samples were expressed differently significantly (Fig. 1
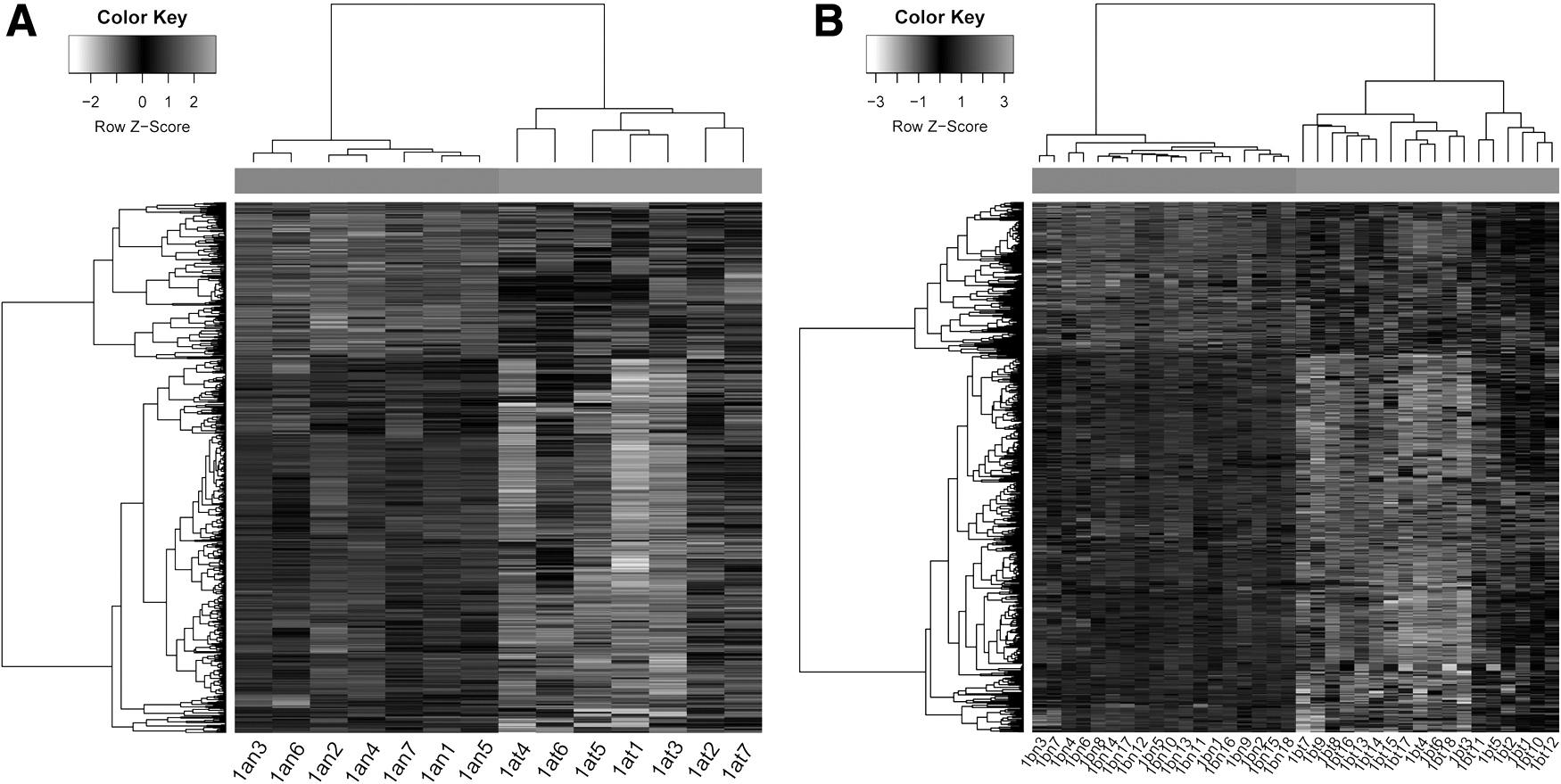
Hierarchical cluster map of DEGs. (
3.2. Functional enrichment analysis
GO term and KEGG pathway enrichment analysis were used to analyze the function of common DEGs (Fig. 2). The upregulated DEGs were significantly enriched in GO terms, including plasma membrane and extracellular region, as well as KEGG pathways such as hypertrophic cardiomyopathy and complement and coagulation cascades cell and adhesion molecules (CAMs) (Fig. 2A). The downregulated DEGs were significantly enriched in GO terms, including microtubule cytoskeketon and spindle pole, and KEGG pathways such as p53 signaling pathway, extracellular matrix (ECM)-receptor interaction, and cell cycle (Fig. 2B).
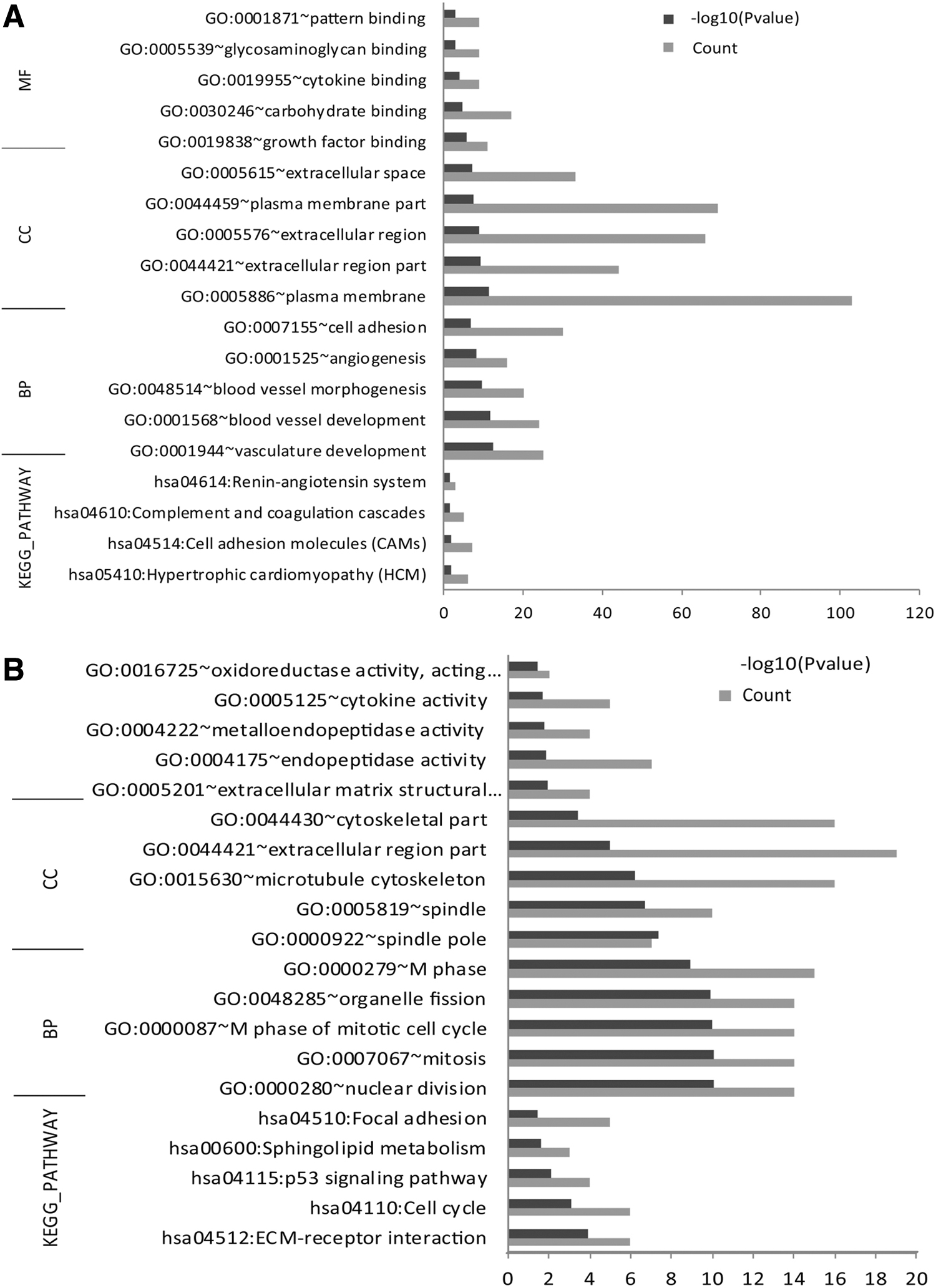
The enriched GO and KEGG pathways for DEGs.
3.3. Hub genes screening from the PPI network
Based on the information of STRING database, the PPI networks were built. As shown in Figure 3, a total of 229 DEGs were included in the PPI network, resulting in 513 matching pairs. Ten hub genes were obtained, including fibroblast growth factor 2 (FGF2), interleukin 6 (IL6), TOP2A, CCNB1, von willebrand factor (VWF), cyclin B2 (CCNB2), budding uninhibited by benzimidazoles 1 (BUB1), matrix metalloproteinase 1 (MMP1), Caveolin-1 (CAV1), and Angiopoietin 1 (ANGPT1), that exhibited especially high degree scores (≥16). There were 3 significant sub-networks selected from the PPI network, including module A (13 nodes, 68 relationship pairs), module B (18 nodes, 33 relation pairs), and module C (17 nodes, 34 relation pairs). All nodes in module A were downregulated genes. In module B, 13 of 18 were upregulated genes, whereas 12 of 17 were upregulated genes in module C (Fig. 4).

PPI network. Dark grey nodes are upregulated genes, and light grey nodes are downregulated genes. PPI, protein-protein interaction.
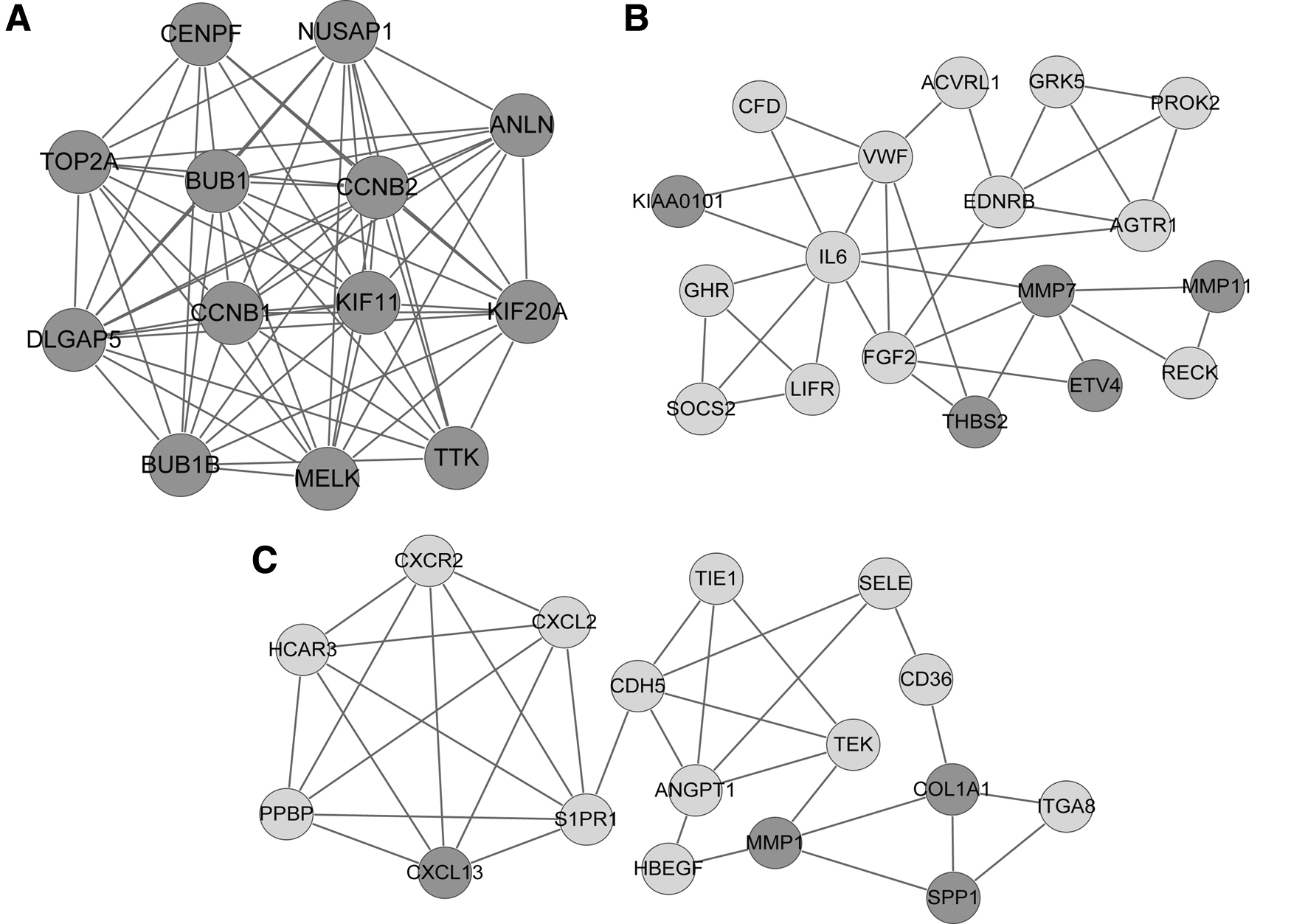
Subnetwork modules of PPI. (
3.4. Analysis of WGCNA modules
Based on the cut-off criteria |log2 (FC)| > 1 and p < 0.05, a total of 3432 DEGs were obtained by comparison of all NSCLC samples with normal samples. Then, we calculated the correlation degree of the DEG and constructed a WGCNA gene co-expression network. Five WGCNA modules were identified from 3432 DEGs as shown in Figure 5. Then, we enriched 20 KEGG pathways from 5 WGCNA modules, MEpink (0 pathway), MEblack (1 pathway: focal adhesion), MEblue (1 pathway: 1 carbon pool by folate), MEcyan (1 pathway: ECM-receptor interaction), and MEgreen (17 pathways: focal adhesion and cytokine-cytokine receptor interaction) (Table 1).
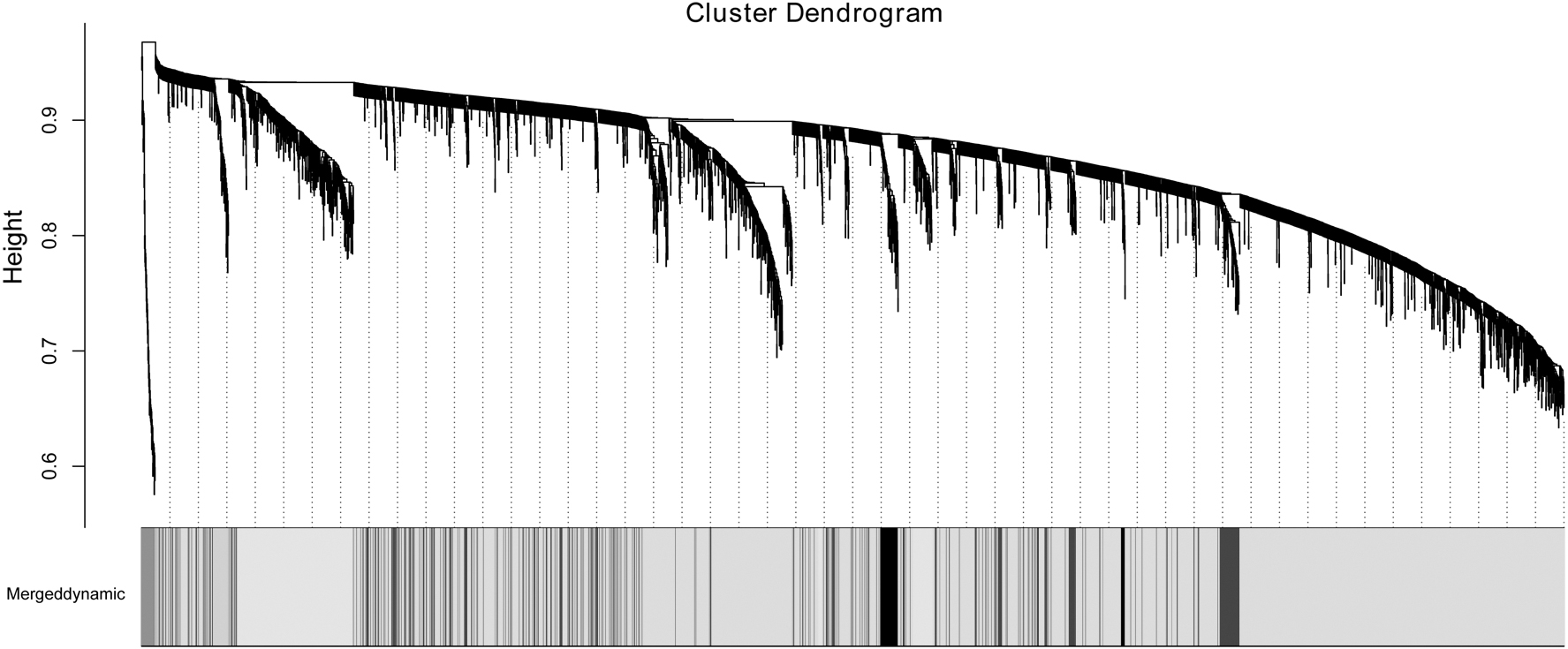
WGCNA co-expression network analysis diagram with different shadings representing different highly co-expressed gene modules. WGCNA, weighted gene co-expression network analysis.
Kyoto Encyclopedia of Genes and Genomes Pathway Enriched by Genes in Five Significant Co-expression Modules
CAM, cell and adhesion molecule; ECM, extracellular matrix.
3.5. Confirmation of the identified DEGs based on LUAD data from TCGA database
With the cut-off values of |log2 FC| > 2 and p < 0.05, a total of 1797 DEGs were revealed in LUAD data from the TCGA database. By intersection analysis of DEGs identified from LUAD data and the common DEGs identified from GSE27262 data, 285 DEGs identified from LUAD data were common DEGs identified from GSE27262 data (Fig. 6A), including TOP2A and CCNB1. Further survival analysis showed that upregulation of TOP2A and CCNB1 was correlated with poor survival of patients (p < 0.05, Fig. 6B, C).
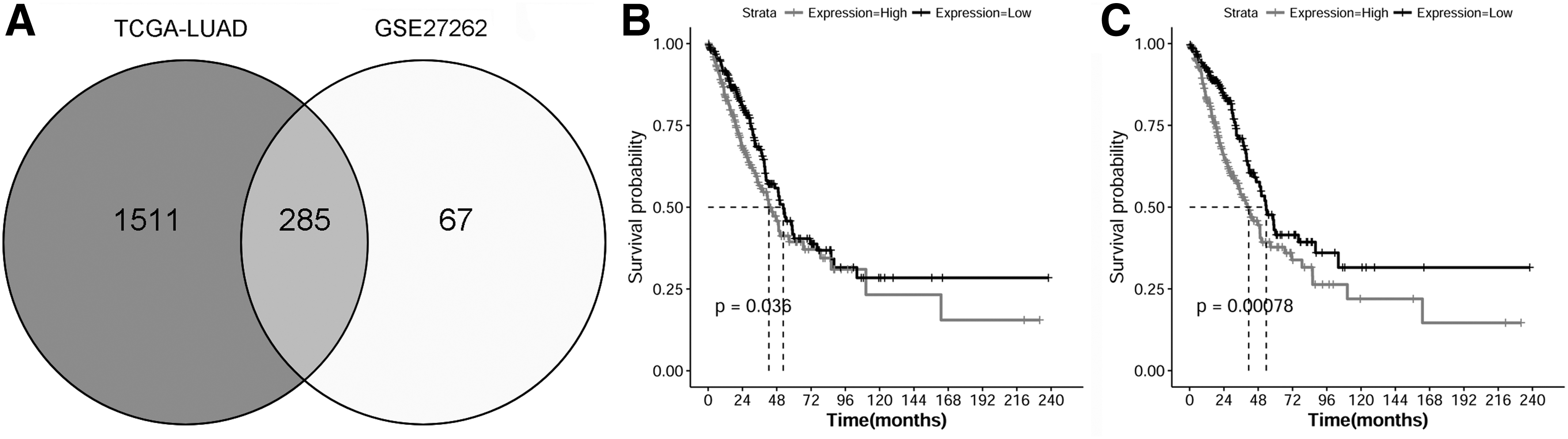
The VENN intersection analysis of DEGs identified from LUAD data and the common DEGs identified from GSE27262 data
4. Discussion
The carcinogenesis, progression, and metastasis of lung cancer are considered very complex processes, because they involve a number of genes and aberrations of pathways (Vogelstein and Kinzler, 2004). To improve the diagnosis and treatment of NSCLC, it is very important to discover abnormal expressed genes and understand their role in NSCLC. With the development of microarrays and high-throughput sequencing, the pathological mechanisms of cancer examine the aberrations at the whole-genome level (Liang et al., 2016).
In this study, 802 DEGs in 1a NSCLC and 734 DEGs in 1b NSCLC samples were obtained, and common DEGs between NSCLC 1a and NSCLC 1b were selected, including 255 upregulated DEGs and 97 downregulated DEGs. Four pathways related to CAMs were enriched in upregulated genes; whereas five pathways related to ECM-receptor interaction, p53 signaling pathway, cell cycle, sphingolipid metabolism, and focal adhesion were enriched in downregulated DEGs. In the module MEgreen analyzed by WGCNA, we found 17 pathways, including focal adhesion, cytokine-cytokine receptor interaction, and purine metabolism pathways. It was important for focal adhesion acting on tumor metastasis, embryonic development defects, and angiogenesis disorder (Yamaguchi et al., 1998). Cytokines are low-molecular-weight (15–30 KD) proteins secreted by a variety of cells, which bind to the corresponding receptors; regulate cell growth and differentiation; and participate in immune, inflammatory response and wound healing (Oppenheim, 2001). Cytokines existing in the body promote or inhibit each other, and they form a very complex network of cytokines (Pierre, 2004). We speculate that focal adhesion and the cytokine-cytokine receptor interaction pathway may be related to NSCLC.
According to Cavallaro and Christofori Ugo and Gerhard (2004), cell adhesion banks, cellular signal transduction, and interactions between cellular environments were closely related to CAMs. In this study, we found that LUAD was mainly concentrated in adhesion junction, complement system, ECM-receptor interaction, and other pathways. Adhesion plaque is a subcellular macromolecule that mediates ECM adhesion to cells (Chen et al., 2003). One of the basic biological mechanisms for maintaining normal functioning was the transmission of information between signaling molecules in multicellular organisms. Some of these signaling molecules are soluble (such as growth factors, hormones, etc.), and others are insoluble (e.g. ECM) (Kinoshita et al., 1995; Chen et al., 2003). All these reports indicate that CAM, cytokine-cytokine receptor interaction, and adhesion junction are involved in the pathogenesis of malignant tumors mainly by affecting cell cycle and mitosis.
The PPI analysis showed that TOP2A and CCNB1 were the hub nodes in the PPI network with the highest degree. Moreover, the survival analysis showed that the upregulation of TOP2A and CCNB1 was correlated with poor survival of patients. The TOP2A gene encodes an enzyme that is involved in DNA replication and cell cycle progression (Resende, 2013). TOP2A has exhibited prognostic importance in various carcinomas, such as prostate cancer Resende (2013) and breast cancer (Brase et al., 2010). Moreover, upregulation of TOP2A is also found to be associated with poor prognosis in LUAD patients (Perumal et al., 2012). In addition, CCNB1 is a crucial cell cycle regulator and G2/M phase promotor. Zhuang et al. (2018) confirmed that upregulation of CCNB1 in tumor tissues could predict the worse survival of hepatocellular cancer patients. A very recent study also reveals that the high expression level of CCNB1 correlates with poorer overall survival in NSCLC patients (Xiao et al., 2018). In agreement with these previous findings, our results prompt us to speculate that key genes, such as TOP2A and CCNB1, may be closely related to the development and prognosis of NSCLC. However, we did not perform more functional experiments to specify their working mechanisms, which could be a possible limitation to our analysis.
The hub genes and key pathways identified in this study are helpful for a comprehensive knowledge of the molecular mechanisms of NSCLC. These findings will provide a theoretical basis for the occurrence of NSCLC and provide biomarkers for the diagnosis and treatment of NSCLC.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
The work was supported by the National Natural Science Foundation of China (No. 81472809, 81502653, 81672983, 81703028), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (No. JX10231801), and the Six Major Talent Peak Project of Jiangsu Province (No. 2013-WSN-040).