
Abstract
Inferring potential associations between microRNAs (miRNAs) and human diseases can help people understand the pathogenesis of complex human diseases. Several computational approaches have been presented to discover novel miRNA-disease associations based on a top-ranked association model. However, some top-ranked miRNAs are not easily used to reveal the association between miRNAs and diseases. This study aims to infer miRNA-disease relationship by identifying a functional module. We first construct a miRNA functional similarity network derived from a disease similarity network and a known miRNA-disease relationship network. We then present an improved K-means (i.e., IK-means) algorithm to detect miRNA functional modules and used 243 diseases to validate the performance of our proposed method. Experimental results indicate that the performance of IK-means is better compared with classical K-means algorithms. Case studies on some functional modules further demonstrate the applicability of IK-means in the identification of new miRNA-disease associations.
1. Introduction
MicroRNAs (miRNAs) are small noncoding RNA molecules containing about 22 nucleotides that play critical roles in the post-transcriptional regulation of gene expression and various biological activities (Ambros, 2004; Li et al., 2017, 2018). miRNA dysfunctions are associated with various complex human diseases (Ambros, 2004; Wang et al., 2014; Zhao et al., 2015; Yang et al., 2016). Consequently, potential associations between miRNAs and diseases should be identified to understand the mechanisms of pathogenicity and to provide valuable references for the diagnosis and treatment of human diseases.
Although numerous technologies, such as microarrays and polymerase chain reactions (Kanno et al., 2006), have been experimentally used to determine relationships between miRNAs and diseases, they have certain limitations, such as high costs and time-consuming operations. Therefore, computational methods should be developed to infer the potential associations between miRNAs and diseases. Numerous computational models have been developed to obtain latent miRNA-disease links based on functionally similar miRNAs that likely interact with similar diseases and vice versa (Zeng et al., 2016; Zou et al., 2016; Luo et al., 2017; Zhao et al., 2018).
Jiang et al. (2010) first introduced a similarity-based method that integrates few data sources consisting of miRNA functional similarity data, experimentally validated disease-miRNA links, and phenotype similarity data to infer potential links between miRNAs and diseases with hypergeometric distribution. However, their method has serious shortcomings with high false-positive and false-negative rates (Yu et al., 2016). Chen et al. (2012) developed a novel method named RWRMDA by executing random walks restarting on a constructed functional similarity network to detect new interactions between miRNAs and diseases. Shi et al. (2013) presented another random walk model to reveal the functional relationship between miRNA targets and disease genes in a protein–protein interaction network. Xuan et al. (2015) used random walk to model the prediction process of the association between miRNAs and diseases on a miRNA functional similarity network. Luo and Xiao (2017) designed a novel method called BRWH to prioritize disease-associated miRNAs on a miRNA functional similarity network constructed from multiple biological data sources. Chen et al. (2016) created a heterogeneous graph-based model named HGIMDA to infer the likelihood of latent links of each candidate disease–miRNA interaction. Some machine learning methods have been introduced to determine potential associations between miRNAs and diseases on a miRNA functional similarity network (Chen and Yan, 2014; Chen et al., 2015; Li et al., 2017).
However, these methods mainly focus on top-ranked association models, which cannot easily reveal latent associations between diseases and miRNAs, such as the association between has-mir-508 and breast neoplasm.
On the basis of previous studies (Cao et al., 2015, 2016, 2018a, 2018b; Liang et al., 2016; Luo et al., 2018), we develop a method called the improved K-means (IK-means), which can effectively mine the latent associations between miRNAs and diseases. This algorithm involves three major steps. First, a clique expansion method is improved to generate the number of clusters. Second, the functional similarity value between miRNA pairs is adopted to replace the distance between nodes, and the IK-means algorithm iteratively updates the clustering center in each clique structure until no changes are observed in the miRNA substructure, which is called the candidate functional module. Finally, the overlapping extent between each pair of candidate miRNA functional modules is quantified. In this step, those candidate functional modules with an overlapping score above the specified threshold are merged, and functional modules with a density less than 0.001 are filtered. Our experiments show that our method achieves satisfactory results in terms of Sn, ACC, and composite score. Case studies are conducted on some miRNA functional modules to further evaluate our proposed algorithm.
2. Materials and Methods
2.1. Method overview
The IK-means algorithm mainly involves three steps (Fig. 1). First, similarity is estimated for each disease pair and miRNA pair, and a miRNA functional similarity network is constructed accordingly. Second, potential associations are inferred. Clique extension is first implemented in the miRNA functional similarity network based on similarities between miRNA pairs to obtain unknown associations between miRNAs and diseases, and the IK-means algorithm is executed on the basis of the identified miRNA cliques to determine the number of functional modules. Similarities between miRNA pairs are used as distances between miRNA pairs. Finally, a number of miRNA functional modules are determined as candidates during the iterative process, and unvalidated miRNAs in functional modules for a given disease are selected as potential disease-related miRNAs.
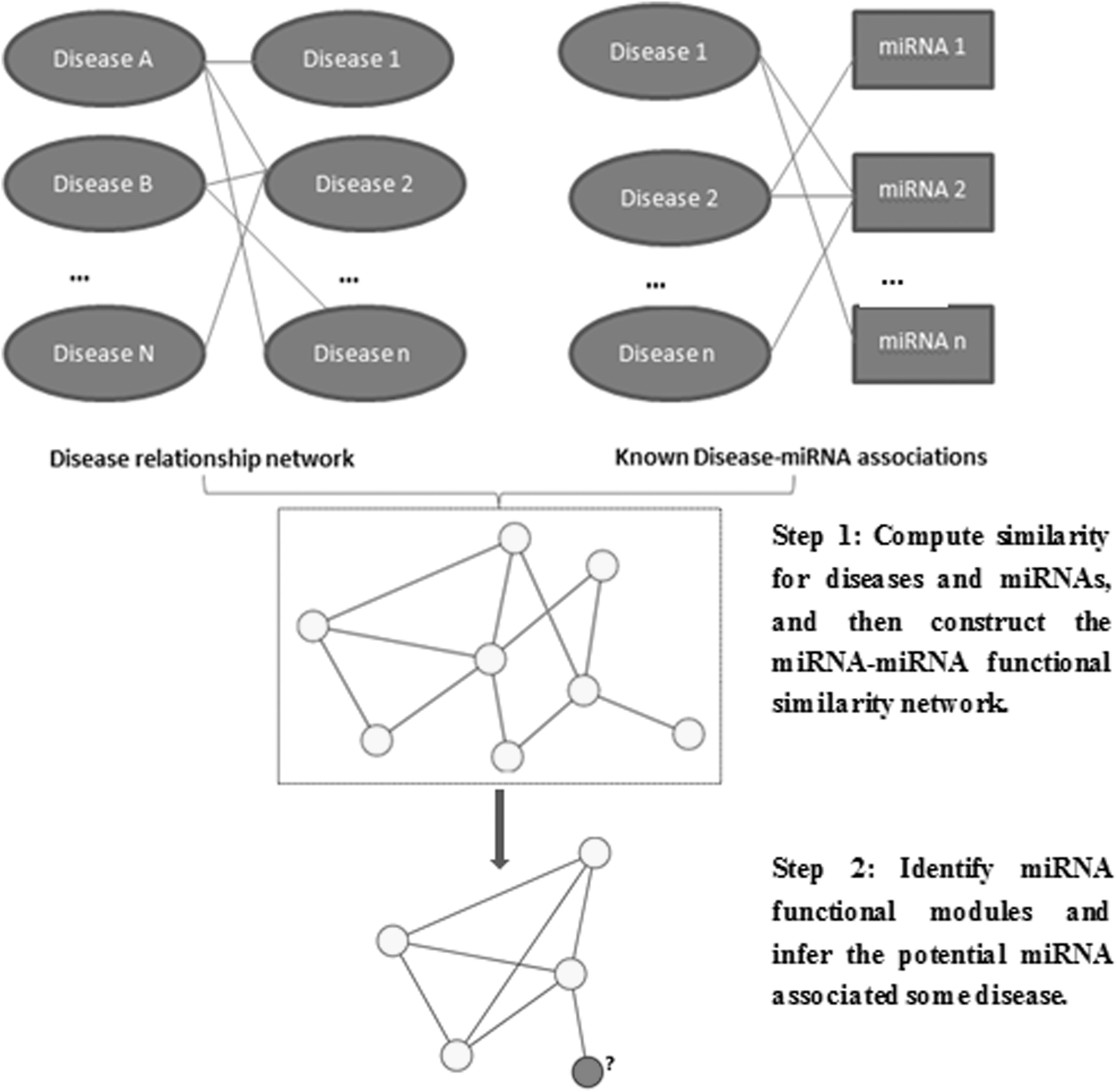
Overall workflow of improved K-means for inferring potential miRNA-disease associations. miRNA, microRNA.
2.2. Constructing a miRNA functional similarity network
2.2.1. Data preparation
The Human microRNA Disease Database (HMDD, released in June 2014) (Li et al., 2014), including experimentally validated miRNA-disease associations, is downloaded. A total of 5100 distinct experimentally confirmed associations between 326 diseases and 491 miRNAs are obtained after duplicate records are removed, and 243 diseases are used as a benchmark dataset after single miRNA-associated diseases are filtered to assess the performance of the IK-means algorithm.
2.2.2. Disease semantic similarity measurement
Let m1 and m2 represent two different miRNAs to construct the miRNA functional similarity network. The functional similarity of miRNAs m1 and m2 is defined as follows (Wang et al., 2010; Luo and Xiao, 2017):
where
where dl denotes a disease, and
where DSim is the disease similarity matrix, and the elements are the similarity values or the corresponding disease pairs.
In this way, the miRNA functional similarity network with 484 miRNAs and 24,061 edges is constructed (Fig. 2).

The miRNA functional similarity network was shown by Cytoscape, and the edges were added if the similarity value of two miRNAs is equal to or greater than 0.4.
2.3. IK-means algorithm
Unlike traditional clustering algorithms (Hartigan and Wong, 1979; Cao et al., 2018a), the IK-means algorithm, which is based on the functional similarity value between miRNA pairs, can effectively infer potential miRNA-disease associations by identifying the functional module. Table 1 describes the IK-means algorithm that involves three steps. First, clique expansion is improved to generate the number of clusters as described in Table 2. Second, the functional similarity value between miRNA pairs is adopted to replace the distance between nodes. Then, starting from clique structures, the improved algorithm iteratively updates the clustering center in each clique structure until no changes are observed in the miRNA substructure, which is called the candidate functional module, as described in Table 3. Finally, the overlapping extent between pairs of candidate miRNA functional modules is quantified. In this step, those candidate functional modules with overlapping scores above the specified threshold are merged, and functional modules with a density of less than 0.001 are filtered. In our study, the overlapping score is set to 0.64. Table 4 describes the merging procedure.
Description of Improved K-Means Algorithm
IK, improved K-means; miRNA, microRNA.
Description of Clique Expansion Algorithm
Description of Clique Center Selection
CCS, Clique Center Selection.
Description of Merge MicroRNA Functional Module
In Table 2, depth-first search and width-first search algorithms are adopted to generate the number of clusters based on the improvement of CPM (Palla et al., 2005). In our study, the number of clique is set to 4, which is consistent with a previous research (Cao et al., 2016).
In contrast to traditional K-means, our proposed algorithm involves the max–min mode to select the cluster center, that is, the cluster center has the greatest similarity value, and the similarity among the cluster centers is the smallest as described in Table 3. The miRNA with the maximum similarity value in the clique structure is regarded as the new cluster center to update the cluster center. Let
where
3. Results and Discussion
In this section, the network properties of the miRNA functional module and the performance evaluation used in our experiment are first analyzed. Then, K-means and IK-means are comprehensively compared. Finally, a case study is examined to show the result of our proposed method.
3.1. Topological analysis
To further understand the miRNA functional similarity network, we analyze the distribution of the average shortest path length and the clustering coefficient. Figure 3 shows the average shortest path length under different neighbor nodes (degree). As the degree increases, the average shortest path length decreases. The average shortest path length of the miRNA functional similarity network is less than 6, which exhibits a feature of complex networks. In Figure 4, a majority of the clustering coefficients of the miRNA functional similarity network are greater than 0.3. This finding demonstrates that the miRNA functional similarity network is another complex network with a small shortest path length and a high clustering coefficient.

The distribution of average shortest path length.
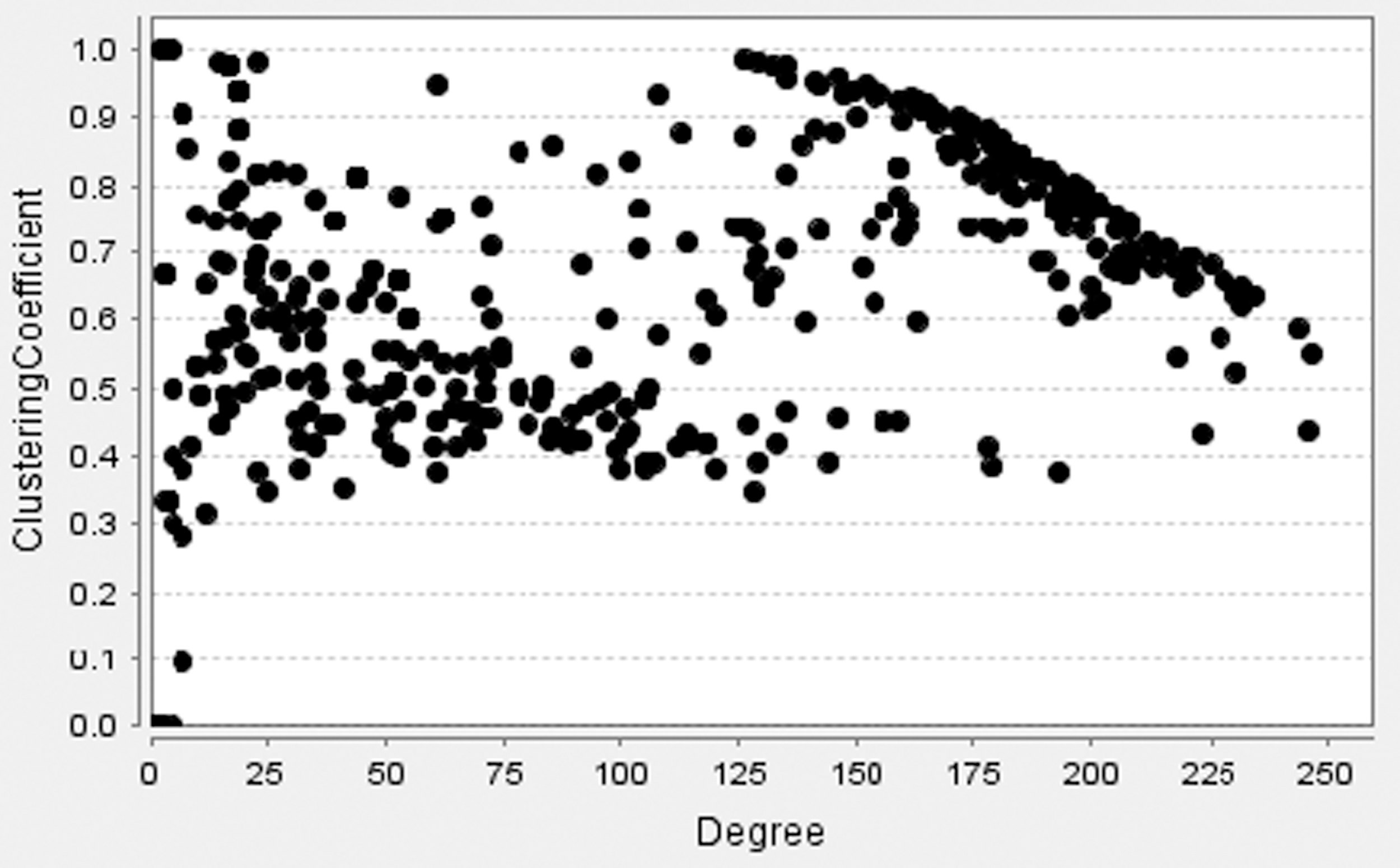
The distribution of clustering coefficient.
3.2. Performance analysis
Before performance analysis is conducted, several evaluation metrics used for comprehensive comparisons are described as follows (Adamcsek et al., 2006; Cao et al., 2016).
F1: F1 is defined as the harmonic mean of precision and recall. This parameter can be used to evaluate the overall identification performance. The corresponding definition is that
Where
Sn: Sn indicates that the detected miRNA functional module covers the number of miRNAs in the benchmark dataset and results from the size of the identified miRNA functional module. A large predicted functional module has a high Sn, which is defined as follows:
ACC: ACC is used to evaluate the quality of detected miRNA functional modules. The corresponding formula is shown as follows:
The composite score of F1, Sn, and ACC is used to analyze the performance of different methods and to highlight the overall performance of our proposed method (Nepusz et al., 2012; Cao et al., 2016).
Table 5 provides the detailed experimental results on the miRNA functional similarity network using 243 diseases as a benchmark dataset. In Table 5, IK-means achieves the highest Sn and ACC. Sn of IK-means is 12.5% higher compared with K-means, and ACC of IK-means is 14.9% higher compared with K-means. Our method achieves a low F1 of 0.153 possibly because some miRNA functional modules generated by K-means match several diseases.
Comparative Results of Improved K-Means with Traditional K-Means Algorithm
The highest value of each data set is shown in bold.
To evaluate the comprehensive performance of these methods, we compare the above methods using the composite score of F1, Sn, and ACC. In Table 5, the composite score of IK-means is 8.15% higher compared with K-means, indicating the effectiveness of our proposed method in inferring miRNA functional modules.
3.3. Case studies
To demonstrate the prediction ability of IK-means in inferring the potential miRNA-disease associations, we analyze case studies and randomly use some functional modules as examples.
IK-means identifies modules (hsa-mir-204, hsa-mir-155, hsa-mir-200b, hsa-mir-223, and hsa-mir-34a), including four miRNAs (hsa-mir-155, hsa-mir-200b, hsa-mir-223, and hsa-mir-34a) associated with esophageal neoplasm (Li et al., 2018). hsa-mir-204 is associated with various diseases, such as lymphoma, prostate cancer, and kidney cancer (www.picb.ac.cn/dbDEMC). On the basis of the above analysis, we can infer that hsa-mir-204 is related to esophageal neoplasm, thereby providing valuable references for biologists.
The disease-associated module (hsa-mir-1181, hsa-mir-431, hsa-mir-448, and hsa-mir-663) is observed using our proposed method. hsa-mir-1181 is associated with and implicated in many cancers, such as colorectal cancer, hepatocellular carcinoma, lung cancer, nasopharyngeal cancer, lymphoma cancer, breast cancer, and ovarian cancer. For example, hsa-mir-1181 is remarkably upregulated in colorectal cancer cells and downregulated in nasopharyngeal cancer cells. hsa-mir-431 is related to brain cancer, lung cancer, breast cancer, malignant sarcoma, ovarian cancer, and other cancers. hsa-mir-448 is linked to some diseases, such as glioma, breast cancer, lung cancer, nasopharyngeal cancer, colorectal cancer, liver cancer, gastric cancer, and kidney disease. hsa-mir-663 is associated with breast cancer, colorectal cancer, lung cancer, prostate cancer, neuroendocrine, and other cancers (www.picb.ac.cn/dbDEMC).
In Section 2.2, we can learn that the functional similarity of each miRNA is calculated on the basis of disease similarity, and the four miRNAs detected by IK-means have high functional similarities. Therefore, we can infer that the functional module consisting of hsa-mir-1181, hsa-mir-431, hsa-mir-448, and hsa-mir-663 may be related to various diseases excluding those diseases validated by databases or literature, which can provide clinical diagnosis for biologists. This finding further demonstrates the effectiveness of our method.
4. Conclusion
Identifying potential miRNA-disease associations can help provide valuable references for clinical diagnosis, treatment, and pharmacy. Existing computational methods have mainly focused on top-ranked models to predict the associations between diseases and miRNAs. However, some top-ranked miRNAs cannot reveal the associations between miRNAs and diseases, such as the association between has-mir-508 and breast neoplasm.
In this study, we proposed a novel method called IK-means to infer miRNA-disease associations based on a functional module. Sn, ACC, and composite score of IK-means were higher than those of the classical K-means algorithm. Case studies on some functional modules demonstrated some potential associations among diseases, indicating that IK-means could reliably reveal miRNA-disease associations for clinical diagnosis.
Despite the effectiveness of IK-means, it has some limitations that should be addressed. IK-means depends on the functional similarity between miRNAs derived from a disease network and disregards the topological structure of a functional similarity network. Considering these limitations, we will integrate the topological structure and the functional similarity network to infer additional potential miRNA-disease associations in our future work.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This work is supported by the National Natural Science Foundation of China grant numbers [61572180, 61472467, 61471164, 61672011, 61862025, and 61602164], the Hunan Provincial Natural Science Foundation of China grant numbers [2016JJ2012 and 2018JJ2024], the Key Project of the Education Department of Hunan Province grant number [17A037], and Jiangxi Provincial Natural Science Foundation of China Grant number [20181BAB211016].