
Abstract
Locus control regions (LCRs), cis-acting, noncoding regulatory elements with strong transcription-enhancing activity, are conserved in sequence and organization, and exhibit strict gene-specific expression. LCRs have been reported and studied in several mammalian gene systems, signifying that they play an important role in eukaryotic gene expression control. Their highly regulated, stable, and precise levels of expression have made them a strong candidate for use in gene therapy vectors. In this study, we attempted to determine the unique signatures of human LCRs by analyzing a data set of LCR sequences for the presence of motifs through systematic bioinformatics approach. Using web-based regulatory sequence analysis tools (RSAT), motif-based analysis was performed. Detected significant motifs were analyzed further for their identity using Tomtom tool. RSAT analysis revealed that significant motifs are existent within the LCRs. Identity analysis using Tomtom showed that detected significant motifs were comparable with known transcription factor (TF) binding sites and the top scoring motifs belong to zinc finger-containing proteins, an important group of proteins involved in a variety of cellular activities. Correspondence to segment of known motif indicates the biological relevance of the detected motifs. Motif-based analysis is valuable for analyzing the various characteristics of sequences, notably TF binding models in this study. Owning to their unique expression control abilities, LCRs form an important component of integrating vectors, therefore identification of unique signatures present within LCR sequences will be instrumental in the design of new generation of regulatory elements containing LCR sequences.
Introduction
Locus control regions (LCRs) are cis-acting DNA transcription regulatory elements that are often involved in the regulation of genes during cellular differentiation and development. Operationally, LCRs are defined by their ability to enhance the expression of linked genes to physiological levels in a tissue-specific and copy number-dependent manner at ectopic chromatin sites (Li et al., 2002). Since the discovery of first LCR, human beta globin locus (HBB-LCR) (Grosveld et al., 1987), several other important LCRs have been discovered in humans and other vertebrates. The most prominent property of the LCRs is their strong, transcription-enhancing activity (Li et al., 2002), and in their absence, the related genes are hardly expressed (Kollias et al., 1986). Enhancer activity of LCRs, in general, resides in the DNAse I hypersensitive sites. These sites help LCRs to form open chromatin structure where DNA is accessible to transcription factors (TFs) that highlight one of the complexities of gene regulation of LCRs. This reinforces the need to study and explore the unique signatures available within the LCRs, notably the TF binding sites (TFBSs).
This study presents the results of motif-based analysis of human LCRs. Sequence motifs, short and recurring patterns in DNA sequence, have become increasingly important in the analysis of gene regulation (D'haeseleer, 2006). Derived sequence motifs, both computationally and experimentally, are useful in deciphering regulatory network of genes, and their mounting value have made them important tools for computational biology in the postgenomic era (D'haeseleer, 2006).
Characterization of the specificities of regulatory sequences is of primary importance to understand the biological roles they play. Computational tools such as regulatory sequence analysis tools (RSAT) (Van Helden, 2003) have become increasingly important for such investigations. RSAT aims at deploying software tools to detect and analyze noncoding regulatory elements in genomic sequences, through a web interface, and over the last 20 years, RSAT suite has maintained uninterrupted services, while extending developments prompted by the advances in the field of regulatory genomics (Nguyen et al., 2018). Motif discovery and motif scanning to predict TFBSs are main functionalities of RSAT.
High-throughput sequencing era has given rise to drastic increase in the number of TF binding models stored in established motif databases such as JASPAR (Khan et al., 2018) and HOCOMOCO (Kulakovskiy et al., 2017). To reveal the identity of unknown and/or detected motifs, motif comparison tools such as Tomtom (Gupta et al., 2007) are available. Tomtom provides a large number of target motif databases with demonstrated accuracy in finding similar motifs. Motif-based analysis is valuable for analyzing the characteristics of sequence motifs such as novel TFBSs. We demonstrate in this study, analysis of data set of human LCR sequences for the detection of significant motifs using RSAT. The detected motifs were further compared and analyzed using Tomtom tool. Motif-based analysis is valuable for analyzing the various characteristics of sequences, notably TF binding models in this study. LCRs forms an important component of integrating vectors, therefore identification of unique signatures will be instrumental in the design of vectors containing LCR sequences.
Materials and Methods
Set of sequences used for motif analysis
Human LCR sequences were retrieved from NCBI database. Upon searching “human locus control region,” 36 gene hits for Homo sapiens were found in the NCBI database. Only 10 sequences of these 36 hits had the feature types of human LCR and/or trans cis regulatory sequence (LCR-like regulatory region) that formed the set of sequences for this study. Description of sequences and their nucleotide accession numbers are given in Table 1. Clinical significance of the retrieved sequences was also collected using the ClinVar link given in the sequence webpage of NCBI.
Summary of the Set of Sequences Used in This Study
Summary of the Set of Sequences Used in This Study
LCR, locus control region.
Overrepresented oligonucleotides in the set of sequences were discovered using RSAT online web server (http://rsat.sb-roscoff.fr/) following the protocol published by Defrance et al. (2008) with minor modifications. In brief, oligo analysis option from the motif discovery menu in the RSAT web tool was used to detect overrepresented oligonucleotides. File containing set of sequences was uploaded and analysis was completed using standard parameters for the organism Homo sapiens. Convert assembled patterns to matrices option was checked/activated before running the program to scan the sequences to build position-specific scoring matrices (PSSMs).
Comparison of the detected motifs using Tomtom
Online web tool Tomtom (Gupta et al., 2007) was used to compare the identified motifs to a database of known motifs. Consensus of the generated matrices of the top assembled patterns were subjected to Tomtom analysis against human database of TF binding models (Kulakovskiy et al., 2017) using the default significance threshold (E-value <10).
Results
Features of the set of sequences
Set of sequences comprised 10 human LCR and LCR-like regulatory region sequences including the relatively known LCR sequences of beta globin (HBB-LCR), alpha globin (HBA-LCR), CD2 (CD2-LCR), and opsin (OPSIN-LCR) along with six other sequences ranging in size from 2261 to 65,247 bp with an average length of 22010.9 bp (Table 1). Variations for the LCR sequences used in this study are present in the ClinVar archive of human genetic variants. Most of the included variations have been interpreted for their clinical significance causing likely benign to pathogenic conditions relative to one or more disorders (Table 1).
Overrepresented oligonucleotides in the set of sequences
Primary result of RSAT analysis displayed a list of overrepresented oligonucleotides sorted by significance in the set of sequences (Fig. 1). Significance of detected oligos is characterized by various attributes in the RSAT result page as listed in the column header of Figure 1. The oligo with the highest occurrence significance value (occ_sig = 23.73) was a hexa-nucleotide “ccccag,” found 355 times in the input set of sequences, overrepresented among the 2080 possible pairs of reverse complements tested for significance. Lower p-value (occ_P = 8.9 × 10−28) indicates the higher significance of the oligo, and also the corresponding expected number of false positives is very low (occ_E = 1.9 × 10−24). Oligos of different oligomer length were overlapping in a manner that was usually a good indication for the relevance of the discovered oligonucleotides.
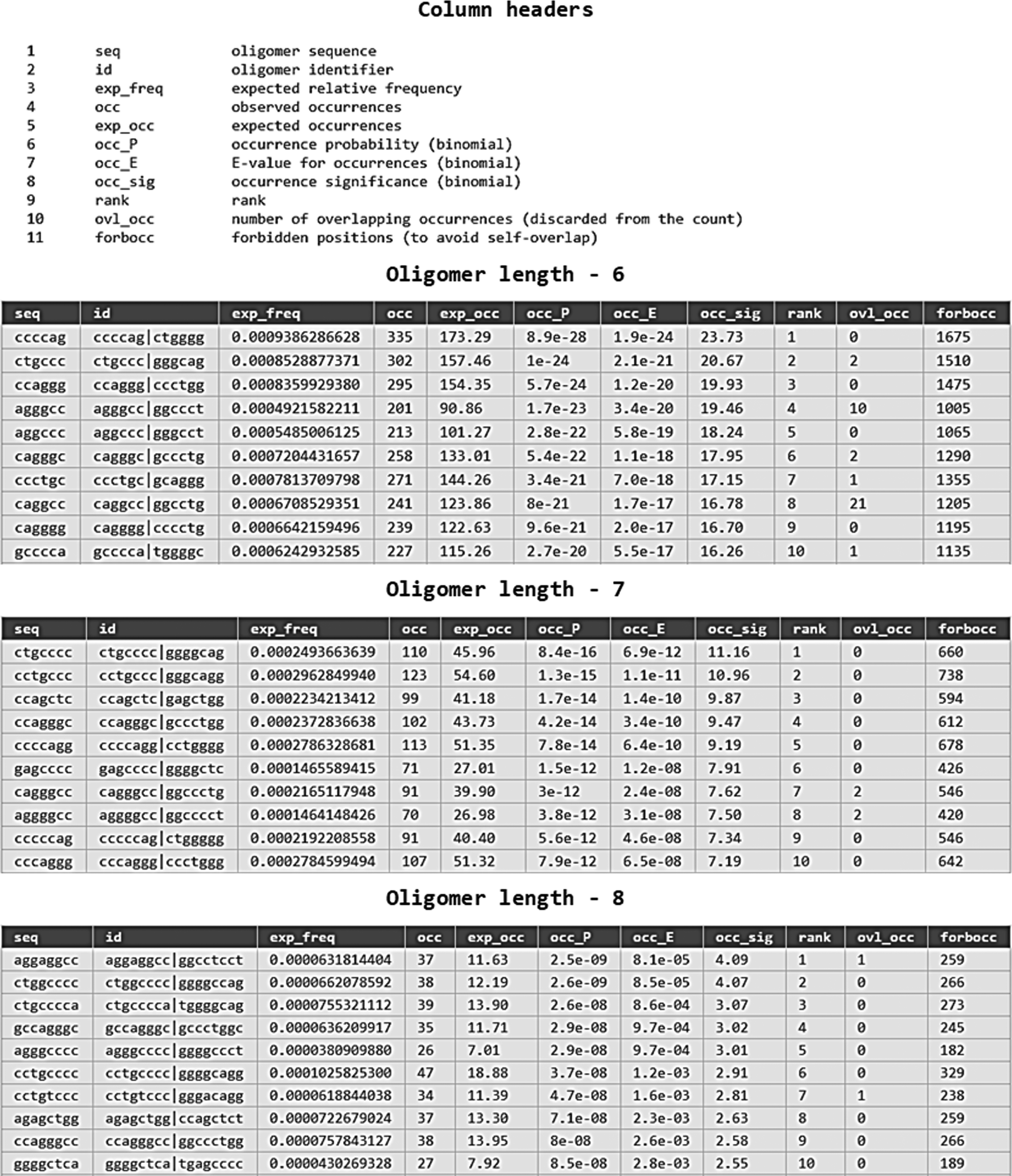
The oligonucleotide tables shows the significant oligos detected in the set of LCR sequences (only top few detected oligos are shown), column header of the oligo analysis indicates the parameters used for the analysis. LCR, locus control region.
Below the primary result, RSAT analysis shows the section pattern assembly, which indicates that several patterns (significant oligonucleotide) could be assembled to generate a larger motif. Several assembled patterns were found in the pattern assembly section, top-ranked assembled patterns are given in Table 2 along with generated matrices and logos of sequence motifs. Top-ranked assembly is a 12-mer (ggagccccagag) for which the top-ranked hexa-nucleotide (ccccag) having highest significance value (23.73) was used as a seed for the generation of this 12-mer. Consensus of generated matrices for this assembly is “ctkgAGCCCCAGAGty.” We had activated the option to convert assembled patterns into PSSMs. An example of generated matrices is given in Figure 2 where the second column of the assembly shows the reverse complement. Numbers in these matrices indicate the highest significance obtained for each residue at each position of the assembly.
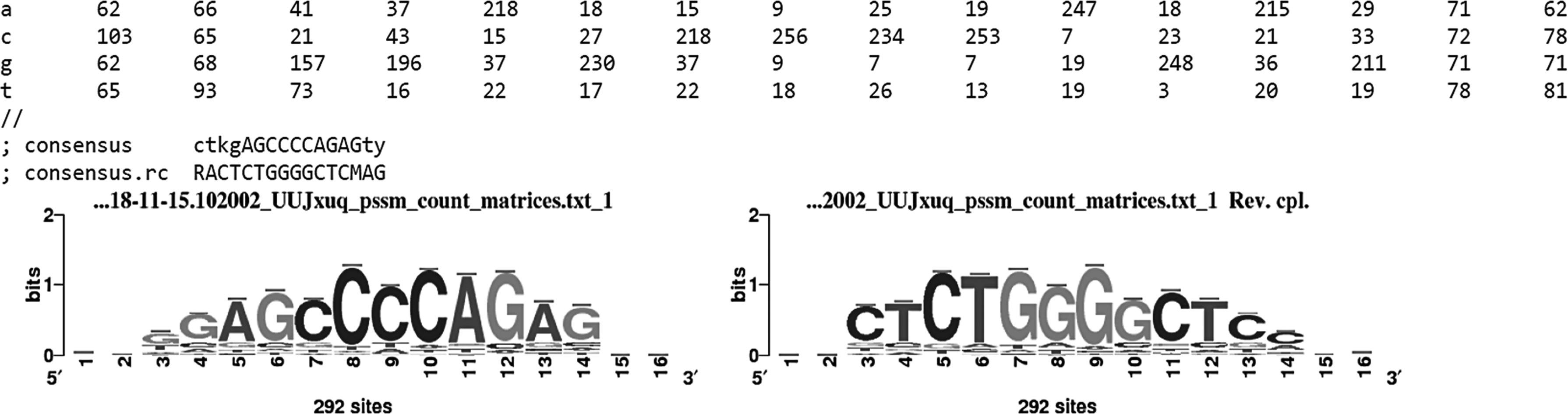
Example of generated matrices; second column of the assembly shows the reverse complement.
Motifs Found by Assembling the Most Significant Oligos Detected in the Set of Locus Control Region Sequences

Consensus of the generated matrices of top assembled patterns as given in Table 2 were subjected to Tomtom analysis to identify any known motifs with similarity to detected motifs after RSAT analysis using a database of human TF binding models. Tomtom identified matching motifs significantly similar to the query motifs (Table 3; E-value <10). A high number of top matching motifs represented the TFs belonging to families of “three-zinc finger Krüppel-related factors,” “more than 3 adjacent zinc finger factors,” and “thyroid hormone receptor-related factors (NR1).”
Top Matching Motifs, Identified Using Tomtom, of Consensus of the Generated Matrices of Top Assembled Patterns
Top Matching Motifs, Identified Using Tomtom, of Consensus of the Generated Matrices of Top Assembled Patterns
TF, transcription factor.
With the discovery of LCRs at different loci of humans and other vertebrates, it is important to study and explore their transcriptional regulation. Motif-based analysis is one approach to identify the essential regulatory elements, notably the binding sites for specific factors within the LCRs. We introduced here motif discovery in the human LCRs, and the discovered motifs were analyzed further for the identification of TF binding models.
Motifs are, in general, short and widespread DNA sequences that play an important role in regulating biological functions. DNA motifs are involved in important processes including determining sequence-specific binding sites for proteins such as nucleases and TF, ribosome binding, mRNA processing, and termination of transcription (D'haeseleer, 2006). Computational methods available nowadays have made it easier to accurately generate putative regulatory sequence motifs by searching for overrepresented patterns in the query sequences that are generally conserved in nature.
Importance of noncoding sequences, for instance LCRs, in transcriptional regulation is being valued by the scientific community worldwide. RSAT is a dedicated tool for the analysis of the noncoding sequences in the genome (Van Helden, 2003). This modular tool detects and analyses cis-regulatory elements in genome sequences through a web interface, motif discovery being one of its main applications (Nguyen et al., 2018). RSAT is a well-documented suite and is unique for its broad range of functionalities and supported organisms from all kingdoms.
We created a data set of 10 human LCR sequences available at and retrieved from NCBI. This data set was subjected to motif analysis using RSAT. Upon RSAT analysis, significant motifs were discovered in the set of LCR sequences. The detected oligos were overlapping in nature, which is a good indication for the relevance of the discovered oligonucleotides, and they generally reveal putative binding sites for exclusive TFs (Defrance et al., 2008). Detected significant oligonucleotides were assembled to generate larger motifs, and assembled patterns were converted into PSSMs that better reflects position-specific variability of the motif. Motif center (corresponding to the most significant oligonucleotide) is highly conserved in the matrix, whereas its flanks are partly degenerated. The matrix can be used to scan new sequences for putative instances of the discovered motif.
Tomtom analysis revealed the identification of motifs by comparing the detected motifs against database of known motifs. Tomtom, a motif database search tool, calculates E-values based on the likelihood of seeing the observed amount of similarity between two motifs by chance, corrected for multiple comparisons typically by using a position weight matrix representation of the motif (Gupta et al., 2007). Web version of Tomtom provides a large number of target motif databases (e.g., JASPAR) where results of query motifs are reported as a list of target motifs, ranked by p-value (Gupta et al., 2007). Top scoring target motifs of the query motifs (consensus of the generated matrices of top assembled patterns) represented the TFs belonging to different families including “more than 3 adjacent zinc finger factors” (e.g., ZFX and GLI3), “three-zinc finger Krüppel-related factors” (e.g., SP1, SP2, and SP3), “factors with multiple dispersed zinc fingers” (e.g., ZN770), “thyroid hormone receptor-related factors (NR1)” (e.g., NR1D1 and RORA), and other TF families.
TFs not only bind to regulatory regions of the DNA but they also interact with other bound TFs and their influence; however, they can be either positive or negative on expression control (Phillips and Hoopes, 2008). Of interest, most of the discovered TFs were zinc finger-containing proteins. These proteins are involved in a variety of cellular activities and constitute the most abundant protein superfamily in the mammalian genome, and are best known as transcriptional regulators (Ravasi et al., 2003). Zinc finger proteins can bind to DNA, RNA, other proteins, or lipids as a modular domain in combination with other conserved structures. Unique features of zinc finger proteins such as high structural stability of the zinc-binding domains makes them perfect for the formation of protein–protein and protein–nucleic acid complexes (Laity et al., 2001). Zinc finger proteins are involved in cell growth regulation and cancer of a large number of tissues (Black et al., 2001). Knockout studies leading to morphological defects very early in development suggest the essentiality of zinc finger proteins during early embryonic development (Marin et al., 1997; Kaczynski et al., 2003).
Increased expression of zinc finger proteins, for example ZFX, is linked with tumorigenesis (Yang et al., 2014; Weng et al., 2015). In a recent study, Rhie et al. (2018) identified 8 nt (AGGCCTAG) motif for ZFX that corresponds to the identified motifs matching ZFX in this study. Rhie et al. (2018) provided insights into the regulation of the cancer transcriptome mediated by ZFX and showed that it acts as a transcriptional activator in multiple types of human tumors. Top assembled motifs matched more than once to other TFs as well belonging to zinc finger protein family, for example SP1, SP2, SP3, and so on. SP factors preferentially bind to motifs containing GC boxes (Thiesen and Bach, 1990; Nagaoka et al., 2001) as the case in this study. SP TFs, particularly SP1, is a highly regulated TF that is involved in regulating expression of a large number of genes that contribute to the “hallmarks of cancer” (Beishline and Azizkhan-Clifford, 2015). In this study, top assembled patterns matching the few important TFs discussed previously on more than one occasion suggests the significance of discovered motifs, having affinity toward TFs implicated in health and diseases, present within the human LCRs.
Motif analysis in this era is an advanced field of sequence analysis in bioinformatics, which is possible because of the high-throughput DNA sequencing technologies such as ChIP-Seq that involves chromatin immunoprecipitation followed by next-generation sequencing to study a plethora of DNA–protein interactions in vivo (Liu et al., 2010). Data generated from such technologies can be utilized to predict motif models using popular web server tools such as RSAT (Van Helden, 2003). Bioinformatics sequence analysis of data generated through sequencing technologies is not only a fine finishing tool for wet-lab results but also provides a source of novel biological knowledge ranging from improved models of TFBS to analysis of specific binding site arrangements (Kulakovskiy and Makeev, 2013). Sequence analysis has also served in finding out actual in vivo binding pattern of particular protein as exemplified by reported motif analysis (Dolfini and Mantovani, 2013). With the development of new computational tools, approaches, and experimental designs, it is now possible to process the immense amount of data already available and being generated at a rapid pace.
Considerable progress has been made toward the development of integrating vectors containing elements of the human LCRs for the treatment of different diseases like sickle cell anemia, β-thalassemia, and severe combined immunodeficiency (Levasseur et al., 2003; Hanawa et al., 2004; Trinh et al., 2009), which prompted us to hunt for unique signatures, notably motifs in this study, present in the human LCRs. In addition, human LCRs are of clinical significance as mutations in the LCR DNA sequence can have a devastating effect resulting in disease (Driscoll et al., 1989).
In conclusion, the results obtained in this study are particularly clean, in that all the top detected significant oligos correspond to segment of known motif indicating the biological relevance of the sites where motifs are located.
Footnotes
Disclosure Statement
The authors declare there are no competing financial interests.