
Abstract
Tuberculosis (TB) is a major public health problem in several countries. Development of first-line and second-line drug resistance strains of Mycobacterium tuberculosis further complicated the management of the disease. Despite available drugs to treat TB, 1.6 million people died from the disease in 2017. In this study, we designed 10 siRNAs against 8 tRNA ligases of M. tuberculosis and validated their usefulness for inhibition of protein synthesis by using computational approach. We found that the predicted siRNAs efficiently form seed duplex complex against their respective mRNA targets. Other different computational approaches were also undertaken to assess the stability, accessibility, and strength of seed duplex complex of designed siRNA and targeted mRNA. On the basis of the computational approach, we reciprocated that the technique will help in opening a new window in the field of TB control program and could be taken for further clinical studies to find their appropriateness for TB eradication.
1. Introduction
Tuberculosis (TB) is a major public health problem worldwide and leading cause of death. Ten million new cases were diagnosed worldwide in 2017, with an estimated mortality of 1.3 million people (World Health Organization [WHO], 2017). TB affects 1 million children and 300,000 deaths were reported among HIV-infected persons. India accounts for the highest number of TB cases (27%) of the total TB notifications in 2017 followed by China (9%) (WHO, 2017). However, the most alarming situation is the emergence of drug-resistant TB. Drug-resistant TB continues to be a public health crisis. In 2017, rifampicin (the most effective first-line drug) resistance was observed in 558,000 TB cases, and of these, 82% had multidrug-resistant TB (MDR-TB) (WHO, 2017). The emergence of even more resistant strains of Mycobacterium tuberculosis known as extensively drug resistant (XDR)-TB is of serious concern. XDR-TB includes the strains of MDR-TB, having resistance to any fluoroquinolone and to at least one of the injectable second-line drugs: kanamycin, capreomycin, or amikacin (Gandhi et al., 2006; Palomino and Martin, 2014).
Molecular diagnostic tools facilitate rapid identification of drug susceptibility status of clinical isolates of mycobacterium tuberculosis (MTB). Drug resistance in MTB isolates is mainly due to the acquirement of single nucleotide polymorphisms (SNPs) in related drug interacting genes. These SNPs alter the protein structure, thus preventing the drugs to bind to the concerning protein for inhibition. These SNPs are also the cause for the upregulation of drug targets, leading to moderate inhibition of the target (Dookie et al., 2018; Wood et al., 2019). Global prevalence of drug-resistant TB is rising continuously (Hanif et al., 2009; Sharma et al., 2011; Sanchez-Padilla et al., 2012; Arnold et al., 2017; Wang et al., 2017; Ismail et al., 2018). Horizontal spread of mono resistance, MDR, and XDR strains of M. tuberculosis in the community further complicates TB management. Thus, there is a pressing need to target new genes with new methods to combat the drug-resistant TB.
Inhibition of target gene expression by antisense sequences can be achieved by several ways but the most common inhibition mechanism is translation repression (Waters and Storz, 2009). Bacterial translation could be reversibly suppressed by trans- and cis-antisense sequences. A single mRNA region may harbor with regulatory regions having cis-antisense sequences and is able to induce sense–antisense intramolecular interactions (Stach and Good, 2011). This sense–antisense intramolecular interactions or folded structure masks ribosomal binding site, preventing translational initiation. A graphical representation of bacterial translation inhibition by siRNA is shown in Figure 1. Bacterial siRNAs are short in length and have stable secondary structure and are resistant to nucleases. The folded secondary structures of bacterial siRNAs have target recognition and binding activity (Majdalani et al., 1998). The folded secondary structure of siRNA may improve target binding through rapid loop–loop interactions. The bacterial siRNAs, involved in antisense interactions, have the “U-turn” structure, which consists of a short hairpin with a tetra-loop containing the YUNR motif (pyrimidine, uracil, any nucleoside, and purine) (Franch et al., 1999). Yanagihara et al. (2005) showed that the inhibition of coagulase enzyme mRNA expression and activity in methicillin-resistant Staphylococcus aureus were achieved by siRNA. Several studies reported the bacterial growth inhibition by antisense RNA for Pseudomonas aeruginosa (Fooladi et al., 2013; Gong et al., 2014) and Lactobacillus paracasei (Ge et al., 2015).
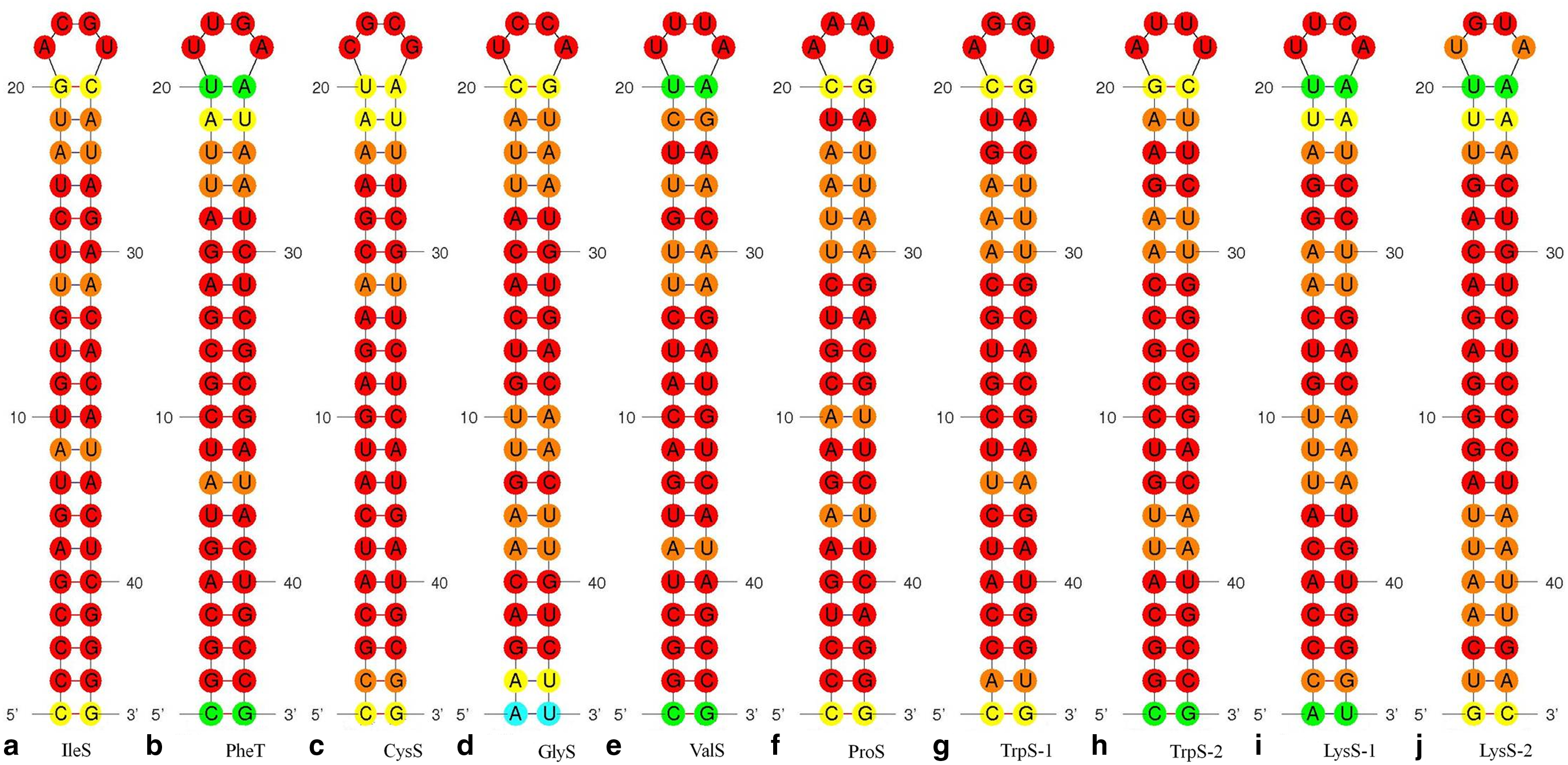
Seed duplex complex formed between predicted siRNA and their mRNA target
Aminoacyl–tRNA synthetase, known as tRNA-ligase, is an essential enzyme that enables the attachment of appropriate amino acid to its tRNA. It catalyzes the esterification reaction of a specific similar amino acid or its precursor to one of all its companionable similar tRNAs to form an aminoacyl–tRNA (Ludmerer and Schimmel, 1987). tRNA-ligase plays an important and central role in RNA translation, expression of genes in protein synthesis, and creation of enzymes to accelerate the metabolic pathways of every form of life. Thus tRNA-ligases are the essential drug/gene silencing targets for inhibition of protein synthesis. In this study, an attempt has been made to design siRNAs against tRNA-ligases of M. tuberculosis and validate their usefulness for inhibition of protein synthesis by using a computational approach.
2. Methods
2.1. Sequence retrieval of tRNA-ligases
The genes of tRNA-ligases of M. tuberculosis (H37Rv) were retrieved from Mycobrowser website (Kapopoulou et al., 2011). All 20 tRNA-ligases were retrieved and used for further analysis. The essentialities of these tRNA-ligases were elucidated by literature search (Sassetti et al., 2003; Griffin et al., 2011; DeJesus et al., 2017).
2.2. Designing of siRNA and secondary structure prediction
Potential siRNAs against tRNA ligases were designed using the siDirect 2.0 online tool (Naito et al., 2009). Ui-Tei, Amarzguioui, Renold (UAR) rules were taken to design nonspecific siRNAs. Melting temperature was set to <21.5°C to allow potential siRNA duplex. All other options were set to default. BLAST was used to check the nonspecific off targets. In first search, nucleotide collection (nr/nt) database was taken with no input in organism option, whereas in the second search, the same was taken with human taxid (taxid:9606) to elucidate the perfect off targets for designed siRNA.
The thermodynamics of interaction between predicted siRNA and target mRNA gene was predicted using RNAcofold program (Bernhart et al., 2006). RNAcofold program calculates the hybridization energy and base pairing form of two RNA sequences. The program gathers extension of McCaskill's partition function algorithm to compute probabilities of base pairing, realistic communication energies, and equilibrium concentrations of duplex structures.
2.3. GC% calculation and secondary structure prediction
GC% of the predicted siRNA was calculated using DNA/RNA guanine and cytosine (GC) content calculator (www.endmemo.com/bio/gc.php). Secondary structure and free energy change for siRNA were predicted using mfold server (Zuker, 2003). Secondary structure formed between mRNA and siRNA guide strands was also predicted using mfold server with default options.
2.4. Heat capacity and concentration plot of duplex calculation
Contributions of each siRNA to the ensemble heat capacity (Cp) and the point at which the concentration of double-stranded molecules of one-half of its maximum value Tm (Conc.) were calculated. Heat capacity plots and concentration plots were calculated using DINAMelt Web server (Markham and Zuker, 2005). The ensemble heat capacity (Cp) is plotted as a function of temperature, with the melting temperature Tm (Cp). Target DNA was converted to the corresponding mRNA and used with the siRNA for calculation of Tm (Cp) and Tm (Conc.).
3. Results
3.1. Designing of siRNA and secondary structure prediction
All 20 tRNA-ligases were retrieved and used for siRNA prediction. Out of 20 tRNA-ligases, siRNAs were predicted in 8 enzymes (Table 1) in accordance with UAR rules. A total of 10 siRNAs were predicted (Fig. 1a–j). tRNA-ligases coding for tryptophanyl-tRNA ligase (TrpS, Rv3336c) and Lysyl-tRNA ligase (LysS, Rv3598c) yielded two siRNAs each whereas the remaining six tRNA-ligases yielded single siRNA. Seed duplex stability temperatures (Tm) for all 10 siRNAs were found in between 6.6°C and 21.9°C (Table 1). The secondary structures for all eight tRNA-ligases mRNA target segments and siRNA guide strands were predicted using mfold server and are shown in Figure 1. The ΔG for predicted secondary structures for target mRNA targets and siRNA guide strands were in between −30.00 and −39.30 kcal. The correlation coefficient for seed duplex formation was calculated as 0.130. Free energy of folding (ΔG) of guide siRNA with mRNA targets was in between 0.00 and 0.50 kcal.
Predicted siRNA Molecules Along with Their Target mRNA, Duplex Stability Temperature (Tm), and GC%
Other thermodynamics details and closing base pair of hairpin loop are given in Table 2. RNAcofold statistics for free energy thermodynamics were found to be equivalent or at par (Table 2) with the free energy values predicted by the mfold server. Frequencies of MFE structure were calculated between 39.84% and 93.42% (Table 2). The partition function folding and dot plots for each of the predicted folded structure were drawn using the RNAcofold server. The dot plots for each of the predicted secondary structure were found to be free from any mutation or SNP, which renders base pairing between target mRNA and predicted siRNA. Only black boxes were noted in the dot plots, indicating the highest integrities of folded structures (Fig. 2). BLAST search results for off target assessment yielded no off targets for each of the siRNA predicted.

Dot plot for predicted secondary structure between siRNAs and mRNA target. Only black boxes were noted in the dot plots indicating the highest integrities of folded structure.
Free Energy Calculations for Each Predicted siRNA Along with Folding Free Energy, Closing Base Pairs, Percentage of MFE Structure, Tm (Cp), and Tm (Conc.)
MFE, minimum free energy. MFE structure of a sequence is the secondary structure that is calculated to have the lowest value of free energy.
3.2. GC% calculation and secondary structure prediction
GC content of siRNA is regarded as a valuable determinant for siRNA and mRNA target duplex formation. All the predicted siRNAs have GC content within 33.33 to 47.61 except tryptophanyl-tRNA ligase (TrpS), which has a GC content of 56.14 slightly less than the reference value 57°C.
3.3. Heat capacity and concentration plot of duplex calculation
The MFE of mRNA secondary structure region targeted by predicted siRNA was calculated to determine the influence of the total number of base pairs in the mRNA region targeted by each siRNA. Moderate to high MFE was observed in all the predicted siRNAs (Table 2). Tm (Cp) for each of the pair was calculated and found in the range of 80.1°C to 87.9°C (Table 2).
4. Discussion
TB is a major health problem worldwide. Nearly one-fourth of the world population is infected with TB. The WHO targets to end TB by 2035 in a phased manner. The goal was set to 35% reduction in TB cases by 2020 in the first phase, 75% reduction by 2025 in the second phase, 90% reduction by 2030 in the third phase, and 95% reduction by 2035 in the fourth phase (WHO, 2018). Development and spread of drug-resistant TB including MDR and XDR-TB are a major concern for the end TB strategy.
Horizontal drug-resistant M. tuberculosis strains were spread in communities by several newly detected TB patients (Basu et al., 2009; Leung et al., 2013; Muller et al., 2013; Eldholm et al., 2015; Shah et al., 2017). These stains are mono resistance to either one of the first-line drugs or MDR strains. Slowly the MDR-TB is reaching the highest magnitude that is a most serious concern. In 2017, rifampicin resistance was observed in 558,000 TB cases, and of these, 457,560 (82%) cases were MDR-TB (WHO, 2017).
Looking at the goal to end TB strategy set by the WHO, there is a need to develop a new strategy to tackle the TB situation effectively. Hence, in this study, we designed and validated the effectiveness of siRNA computationally. For this purpose, tRNA-ligases of M. tuberculosis were targeted for gene silencing through the siRNA technology.
tRNA-ligases are enzymes that enable the attachment of appropriate amino acids to its tRNA at the time of protein synthesis in the translation process. This feature makes this enzyme central to all metabolic pathways. Thus inhibiting tRNA-ligases by hybridizing siRNA would be able to stop translation in M. tuberculosis, ultimately leading to inhibition of the organism.
Melting temperature (Tm) and standard free energy change (ΔG) for the formation of the seed duplex are considered as good measures for the thermodynamic stability of the seed duplex (Ui-Tei et al., 2008). The calculated Tm for 10 seed duplex complexes varied from 6.6°C to 21.9°C, which were negatively correlated with the calculated ΔG values ranging from −30 to −39.30 k/cal. The correlation coefficient was 0.130, indicating a very strong correlation between the seed duplex complex. Thus the designed siRNAs were stable enough to form seed duplex complexes with the target mRNA. The stability of guide strand structure was measured to understand the accessibility of target mRNA and the minimum free energy of folding was varied from 0.0 to 0.50 k/cal, indicating the stability of the guide strand structure. Negative free energy of folding has lower accessibility to target mRNA (Nur et al., 2015). No potential off targets were detected for any of the designed siRNAs. Although some off targets were noticed for human genomic DNA database, those were nonspecific and had no 3′ or 5′ accessibility to the siRNA.
The frequency of the MFE structure in the ensemble of secondary structures and the diversity of the ensemble denote the strength of the base paring pattern for seed duplex complex (Zuker, 2003). A strong seed duplex strength is associated with >66% frequency of the MFE in the ensemble of secondary structures. In this study, the frequencies of MFE ensembles were between 66.37% and 93.42% for all designed siRNA and mRNA seed duplex complexes, indicating the strong association between them.
GC content of siRNA is a major determinant of formation of seed duplex complex between siRNA and target mRNA. At the time of designing of siRNAs, GC-rich target sites should be avoided because of their potential to be involved in strong bonding in their secondary structure (Heale et al., 2005). Lower GC content leads to stronger inhibitory effects, and 30%–57% of GC content is considered sufficient for formation of a strong seed duplex complex between siRNA and target mRNA (Liu et al., 2013). In this study, all the predicted siRNAs have GC content within 33.33 to 47.61, except tryptophanyl-tRNA ligase (TrpS), which has a GC content of 56.14, indicating the idealness of designed siRNA.
Heat capacity temperature Tm (Cp) and the point at which the concentration of double-stranded molecules is one-half of its maximum value Tm (Conc.) are two important determinants for formation of a strong seed duplex complex between siRNA and targeted mRNA. Higher the value of Tm (Cp) and Tm (Conc.), stronger the bonding association between siRNA and targeted mRNA (Shah et al., 2017). In this study, the higher values of Tm (Cp) and Tm (Conc.) showed the stronger seed duplex complex between designed siRNA and targeted mRNA.
5. Conclusions
With the advent of siRNA technology in gene silencing, it is possible to inhibit the expression of different genes in various biological systems. Designing of siRNAs against a particular mRNA and their effectiveness in forming seed duplex complex can also be assessed by bioinformatics tools. In this study, 10 siRNAs were designed against tRNA-ligases of M. tuberculosis using computational methods. Other different computational approaches were also implemented to assess the stability, accessibility, and strength of seed duplex complexes of designed siRNAs and targeted mRNA. In this analysis, we present the designing of 10 siRNAs and their interactions with respective mRNA targets through the RNA folding computational approach. The predicted siRNAs will help in opening a new window in the field of TB control program and could be taken for further clinical studies to find their appropriateness for TB eradication.
Footnotes
Acknowledgment
We are highly thankful to JALMA administration for logistic support.
Author Disclosure Statement
The authors declare they have no competing financial interests.