
Abstract
Approximately half of the unexplained recurrent spontaneous abortions remain unexplained (URSAs). We aimed to provide novel insights into the biological characteristics and related pathways of differentially expressed genes (DE-genes), DE-methylated genes, and DE-miRNAs in URSA, and construct a molecular miRNAs–mRNAs network. Four data sets (GSE22490, GSE121950, GSE73025, and GSE43256) were gained from GEO data sets. We identified the DE-genes, DE-methylated genes, and DE-miRNAs using the LIMMA package in R software. Function and enrichment analyses were conducted using DAVID. A protein–protein network was performed by STRING. We predicted the target genes of DE-miRNA using DIANA-microT-CDS. Then, we constructed miRNAs–mRNAs network. There were 137 genes that overlapped in two expression profile data sets (GSE121950 and GSE22490). We found 10 overlapping DE-methylated genes and DE-genes with opposite expression alteration trends. All those 10 genes were hypermethylated lowly expressed genes. Pathway analysis illustrated that DE-genes were enriched in osteoclast differentiation, leishmaniasis, NF-kappa B signaling pathway, Toll-like receptor signaling pathway, and tuberculosis. Based on protein–protein interaction analysis, TLR8, TLR2, CD86, TLR4, IL10, CD163, FCGR1A, CXCL8, FCGR3A, HCK, PLEK, and MNDA were identified as hub genes for DE-genes. We screened out 47 DE-miRNAs and 42 overlapping DE-genes between predicted target genes of DE-miRNAs and the 137 DE-genes. We then constructed miRNAs–mRNAs network. This study identified several genes and miRNAs involved in the development and progression of URSA, including FCGR1A, FCGR3A, CXCL8, HCK, PLEK, IL10, hsa-miR-498, and hsa-miR-4530. Although further in vivo and in vitro validations are required, our results may provide a theoretical basis for future studies.
Introduction
Approximately 1% of couples attempting to conceive experience recurrent spontaneous abortion (RSA), which is defined as the failure of two or more consecutive clinical pregnancies before 20 weeks of gestation (Rai and Regan, 2006). The etiology of RSA is complicated and multifactorial. Although researchers have identified some causes of RSA, including fetal chromosomal abnormalities, maternal immune and endocrine factors, reproductive tract abnormalities, genital tract infections, and environmental risk factors (Li et al., 2002), approximately half of these cases remain unexplained (unexplained recurrent spontaneous abortion, URSA) (King, 2000).
Decidua, as an important part of the maternal–fetal interface, plays an important role in the establishment and maintenance of pregnancy. Decidual tissue can limit the invasion degree of trophoblast cells, on the one hand to ensure sufficient blood supply for the fetus from the mother, and on the other hand to avoid damage of the mother uterine due to excessive invasion of trophoblast cells. Lipid droplets, glycogen, and growth factors secreted by decidua cells can provide nutrition for embryo and affect its growth and development. Decidualization is accompanied by the formation and remodeling of a large number of blood vessels, which provide sufficient blood supply and nutrition for the growth and development of embryos (Okada et al., 2018). In addition, decidual tissue is also involved in the coordination of maternal and fetal immune responses to protect the fetus from maternal rejection and maintain a normal pregnancy (Norwitz et al., 2001). Failure of cell invasion during early pregnancy, abnormal immune tolerance, and arterial remodeling in the developing decidua, which are controlled by complicated genetic and epigenetic modification network, may lead to URSA (Krieg et al., 2012).
Epigenetic modifications, including DNA methylation, noncoding RNA, genomic imprinting, and histone modification, refer to the heritable changes in gene functions without changing the genetic genes. Abnormal DNA methylation was found in the decidual chorionic villi of URSA with normal karyotype, especially at the loci of the imprinting genes (Hanna et al., 2013; Zheng et al., 2013; Yu et al., 2018). Aberrant microRNAs (miRNAs), which are endogenous small noncoding RNAs and ∼22 nucleotides, were found in URSA (Dong et al., 2014; Wang et al., 2016).
In this study, we used bioinformatics to explore the differentially expressed genes (DE-genes), differentially methylated genes (DE-methylated genes), and differential expressed miRNA (DE-miRNAs) in chorionic villi of URSA patients through analyzing the results of microarray and high-throughput sequencing. We aimed to provide novel insights into the biological characteristics and related pathways of DE- genes, DE-methylated genes, and DE-miRNAs in URSA and construct a molecular miRNAs–mRNAs network.
Methods
Microarray data
The mRNA, miRNA, and methylation genes expression profile data sets of decidua villa in URSA and selective abortion of normal pregnancy were retrieved from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo/). The key words for search included “recurrent spontaneous abortion,” “recurrent pregnancy loss,” “decidual,” “villi,” and “homo sapiens.”
Four data sets (GSE22490, GSE121950, GSE73025, and GSE43256) were gained for further analyses (Fig. 1). The GSE22490 data set and the GSE121950 were used to identify DE-mRNAs. We identified DE-methylated genes between URSA samples and normal control samples by analyzing GSE43256. DE-miRNAs were identified by analyzing the GSE73025 data set.
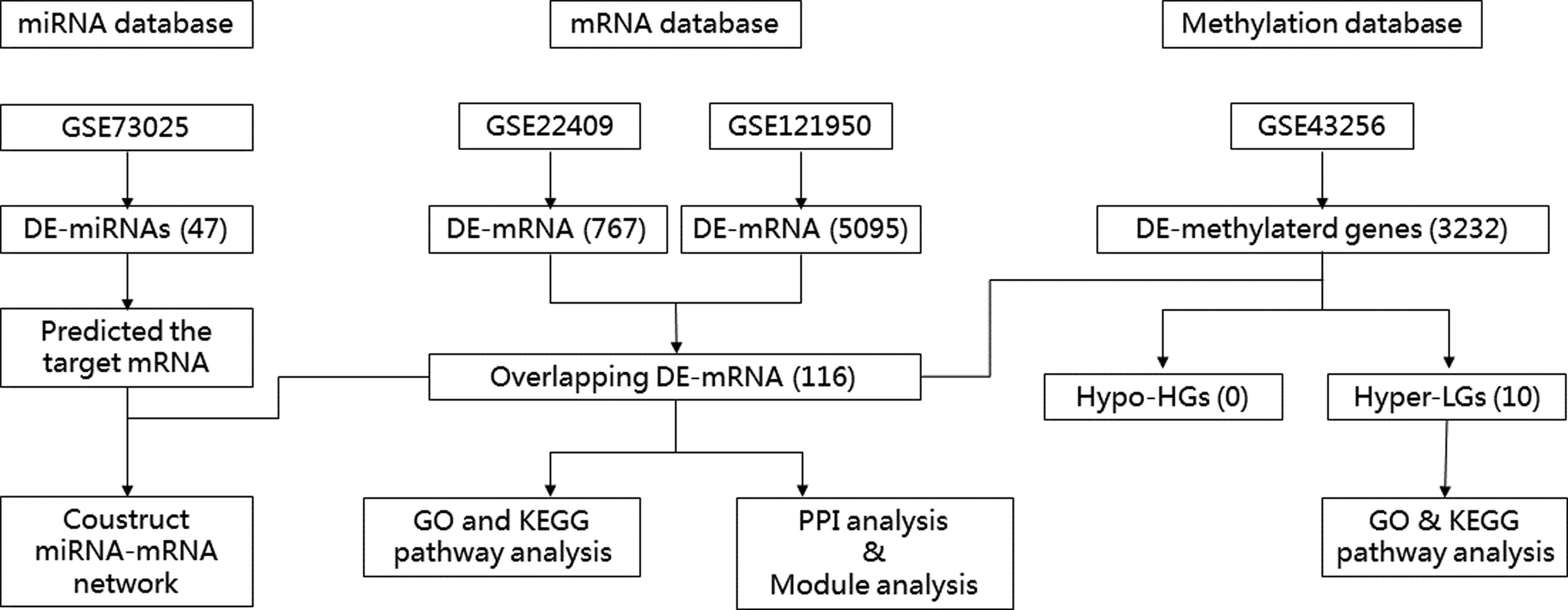
Flow chart of data processing and analysis.
DE-genes, DE-miRNAs, and DE-methylated genes between URSA and normal control were identified using the LIMMA package (Linear Models for Microarray Data) in R software. To detect the DE-mRNA and DE-miRNAs, the p-value <0.05 and |logFC| > 1 cutoff criterion were obtained in the screening. For DE-methylated genes, the threshold was p < 0.05 and |logFC| > 0.01.
Identify overlapping DE-genes and DE-methylated genes
Overlapping downregulated co-DE-genes in GSE22490 and GSE121950 and hypermethylation genes in GSE43256 were identified as hypermethylated lowly expressed genes (Hyper-LGs); similarly, overlapping upregulated co-DE-genes and hypomethylation genes were considered hypomethylated-highly expressed genes (Hypo-HGs).
Functional and pathway enrichment analysis
Annotation on the overlapping DE-genes in GSE22490 and GSE121950 related to URSA was performed using DAVID (http://david.abcc.ncifcrf.gov/) to describe the gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway (Huang et al., 2009). GO analyses included molecular function, biological process (BP) and cellular component. We defined a p value <0.05 as statistically significant for GO terms and KEGG pathways.
Protein–protein interaction network generation and module analysis
The protein–protein interaction (PPI) network was analyzed by STRING (https://string-db.org/) (Lin and Lin, 2017). The cutoff standard was defined as >0.4. We visualized the PPI network by Cytoscape. Hub genes were ranked by Cytoscape cytoHubba (Chen et al., 2014). Cytoscape MCODE was applied to screen modules within the PPI network (Chen et al., 2014). An MCODE score >4 and number of nodes >5 were taken as the criteria to define a module.
Construction of the miRNAs–mRNAs network
Target genes of DE-miRNAs were predicted using DIANA-microT-CDS (www.microrna.gr/microT-CDS) (Liu et al., 2019). The target genes of each DE-miRNA should be differentially expressed in both GSE22490 and GSE121950 data sets with consistent expression trends. Also, the target genes and the miRNA should be differentially expressed with opposite expression alteration trends. The regulatory network of miRNAs-mRNAs was constructed using Cytoscape (www.cytoscape.org/). We used Cytoscape centiscape 2.2 to analyze the network (Liu et al., 2019).
Results
Identification of DE-genes
We found 5095 and 676 DE-mRNAs in the GSE121950 and GSE22490, respectively. There were 137 genes that overlapped. Compared with normal pregnancy, 116 genes were upregulated and 21 genes were downregulated in URSA.
Functional enrichment and KEGG pathway analysis of overlapping DE-genes
For overlapping DE-genes in GSE121950 and GSE22490, we listed GO and KEGG pathways in Figure 2. The most significantly enriched BPs were immune response, phagocytosis, inflammatory response, B cell receptor signaling pathway, and innate immune response. The KEGG pathway analysis showed that DE-genes were most significantly enriched in osteoclast differentiation, leishmaniasis, NF-kappa B signaling pathway, Toll-like receptor signaling pathway, and tuberculosis.
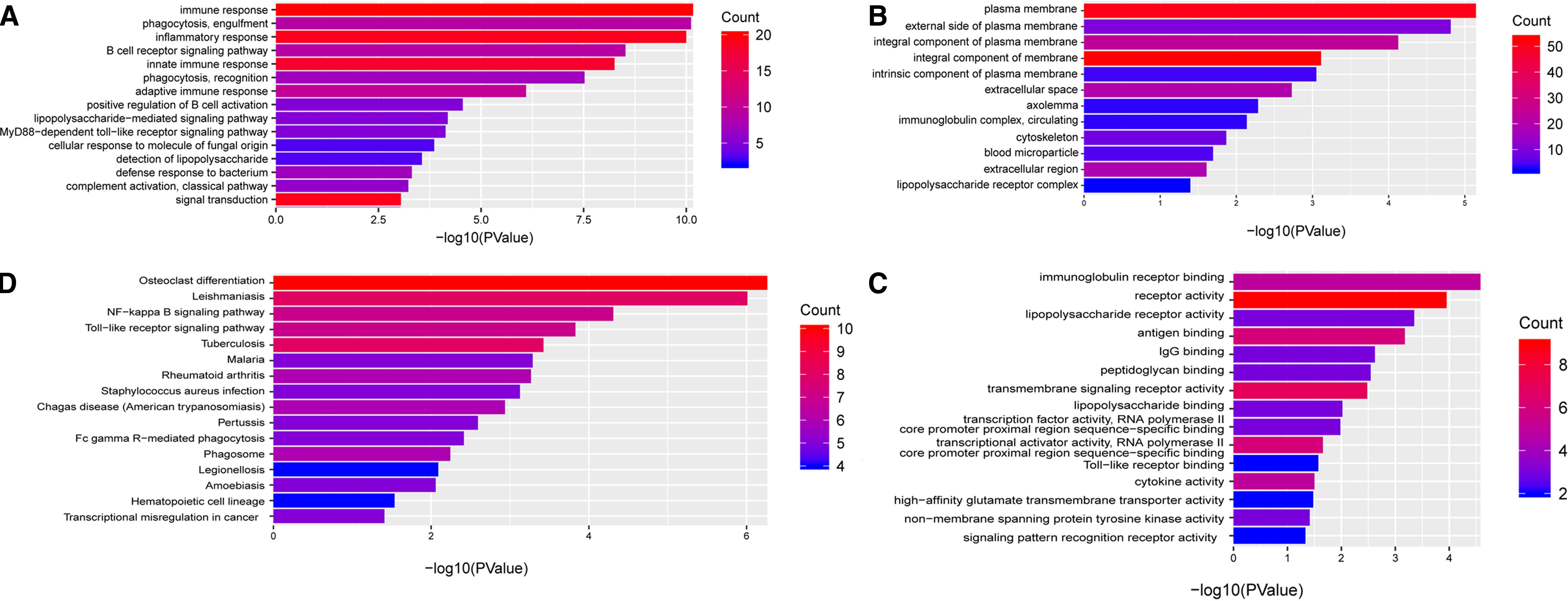
Enriched GO and KEGG pathways analysis of codifferentially expressed genes in GSE22490 and GSE121950 data sets
PPI network of 137 overlapping DE-genes in GSE121950 and GSE22490 was established by STRING, which comprises 127 nodes and 421 edges. TLR8, TLR2, CD86, TLR4, IL10, CD163, FCGR1A, CXCL8, FCGR3A, HCK, PLEK, and MNDA were identified as hub genes by overlapping the top 15 genes according to degree and maximal clique centrality methods in cytoHubba.
Two clusters from the PPI network were established as statistically significant (Fig. 3). Module 1 comprises 20 nodes and 87 edges, and module 2 comprises 12 nodes and 36 edges. Enrichment analyses for module 1 demonstrated that the pathways were mainly associated with leishmaniasis, osteoclast differentiation, tuberculosis, NF-kappa B signaling pathway, and Toll-like receptor signaling pathway. The most significantly enriched BP in module 1 was immune response. The KEGG pathways in Module 2 were mainly rheumatoid arthritis, intestinal immune network for IgA production, leishmaniasis, pertussis, and NF-kappa B signaling pathway. The most significantly enriched BPs in Module 2 included regulation of immune response, positive regulation of immune system process, inflammatory response, cellular response to cytokine stimulus, and positive regulation of immune response.
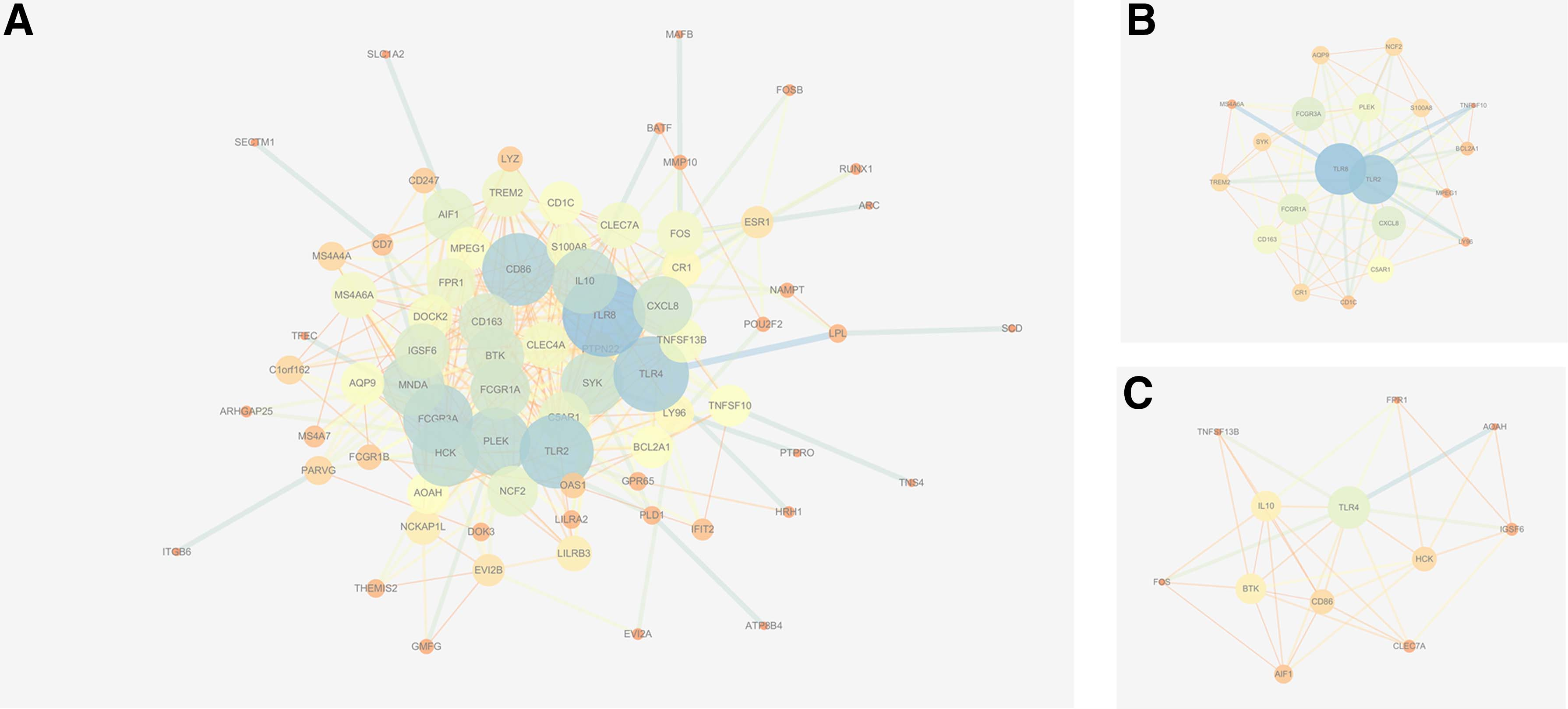
PPI analysis.
We identified 3232 DE-methylated genes, with 1693 hypermethylated genes and 1539 hypomethylated genes. We found 10 overlapping DE-methylated genes and DE-genes with opposite expression alteration trends. All those 10 genes were Hyper-LG, and they were LILRA2, PLEK, EVI2A, GMFG, ESR1, MMP10, PDLIM4, POU2F2, IL10, and TLR4. The BPs were enriched in regulation of nitric oxide biosynthetic process, regulation of Toll-like receptor signaling pathway, positive regulation of platelet activation, regulation of response to external stimulus, and regulation of reactive oxygen species metabolic process. The most significantly enriched KEGG pathways were malaria, inflammatory bowel disease, leishmaniasis, pertussis, and amoebiasis.
Construction of the miRNAs–mRNAs network
We then screened out 47 DE-miRNAs from GSE73025. Among them, 29 miRNAs were upregulated and 18 miRNAs were downregulated. There were 42 overlapping genes between predicted target genes of DE-miRNAs and the DE-genes in GSE121950 and GSE22490. These genes were related to 22 miRNAs. The relationships between mRNAs and miRNAs are shown in Figure 4. hsa-miR-498 (degree = 10) and hsa-miR-4530 (degree = 4) were the most significant miRNAs. hsa-miR-498 negatively regulated BTK, NAMPT, CD86, FCGR1A, LMO3, NCF2, ESR1, IQCH, and MS4A7. hsa-miR-4530 negatively regulated multiple genes, including SLC1A2, ALDOB, SCD, and TENM4.
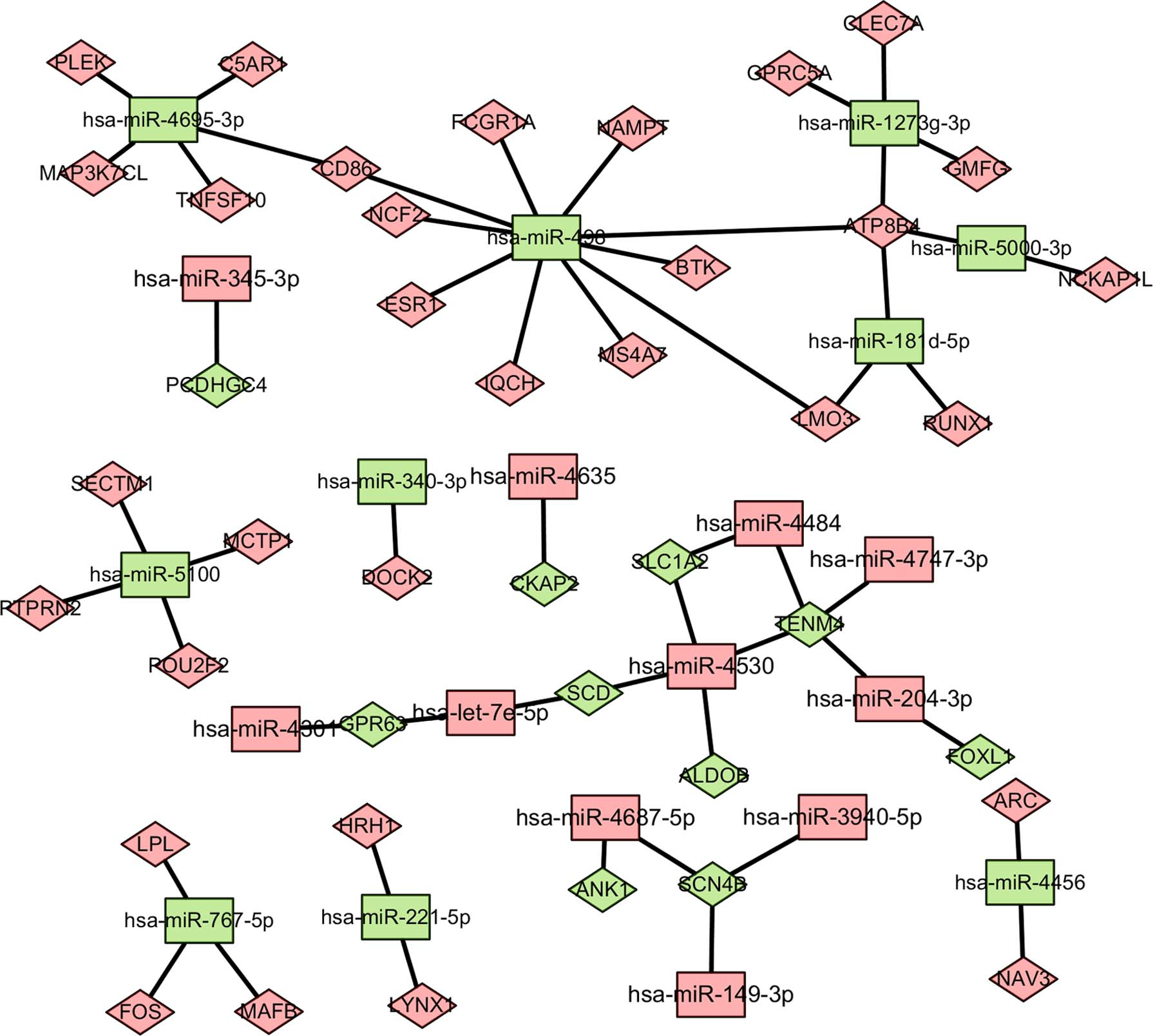
miRNA–mRNA regulation network. Diamond-shaped nodes represent target genes, and rectangular nodes represent miRNAs. Green represents downregulation and red represents the coupregulation.
The occurrence and development of URSA are a complex process that involves genetic and epigenetic disorders. In this study, we identified 137 overlapping DE-genes in two genes expression data sets of decidua villa between URSA and selective abortion of normal pregnancy. Among these genes, 116 genes were upregulated and 21 genes were downregulated in URSA. The most significantly enriched BPs were immune response, phagocytosis, inflammatory response, B cell receptor signaling pathway, and innate immune response. According to PPI analysis of DE-genes, TLR8, TLR2, CD86, TLR4, IL10, CD163, FCGR1A, CXCL8, FCGR3A, HCK, PLEK, and MNDA were identified as hub genes.
TLRs are widely expressed at the maternal–fetal interface, including in immune cells, trophoblasts, and decidual cells. They play an important role in various pregnancy complications, such as abortion, preterm labor, preeclampsia, and even fetal complications (Koga et al., 2014). In early pregnancy loss, TLR stimulation was found to induce fetal resorption (Shimada et al., 2003; Lin et al., 2006). TLR8 was reported to be involved in conceptus development and establishment of pregnancy in sheep (Ruiz-González et al., 2015). TLR2 and TLR4 were considered as the principal receptors for the recognition of bacterial cell wall components at the maternal–fetal interface (Koga et al., 2014).
CDs are membrane proteins mainly expressed on leukocytes. A small number are also expressed on endothelial cells, erythrocytes, and stem cells. CD86 is one kind of receptor involved in the costimulatory signal essential for T-lymphocyte proliferation and IL-2 production, by binding CD28 or CTLA-4. A decrease in the CD86 and CD60 expression and an increase in the CTLA-4 expression were found in the abortion-prone mating (Zhou et al., 2008). CD163 at the fetal–maternal interface was implicated for viral entry into the cell (Novakovic et al., 2016). Genome editing of the CD163 gene through CRISPR/Cas9 prevented porcine reproductive and respiratory syndrome virus infection and reproductive losses associated with infection (Whitworth and Prather, 2017).
Interleukins (ILs) are a group of cytokines that were first seen to be expressed by leukocytes. IL-1 is the first member of the IL-1 family that is closely linked to the innate immune response. IL-1 affects virtually all cells and organs and is a major pathogenic mediator of autoinflammatory, autoimmune, infectious, and degenerative diseases. Low IL-10 levels were reported to be associated with pregnancy complications such as RSA, preterm delivery, premature rupture of fetal membranes, preeclampsia, and intrauterine fetal growth retardation (Azizieh and Raghupathy, 2017). CXCL8, also known as IL-8, is important in inflammation-related diseases, and might play an important role in URSA (Huang et al., 2018).
FCGR1A is a high-affinity Fc-gamma receptor, and it functions in both innate and adaptive immune responses (Brandsma et al., 2017). Hemopoietic cell kinase (HCK) is a member of the sarcoma (SRC) family of cytoplasmic tyrosine kinases. It is expressed in cells of the myeloid, B-lymphocyte cell lineages, and many cancer cells. It enhances the secretion of myelin growth factor and proinflammatory cytokines (Poh et al., 2015). Pleckstrin (PLEK) is a major substrate of protein kinase C in platelets and leukocytes with an important role in exocytosis (Jackson et al., 2011). Besides, phosphorylated PLEK could increase the secretion of proinflammatory cytokine in mononuclear phagocytes (Ding et al., 2007). Myeloid nuclear differentiation antigen (MNDA) is a member of the interferon-regulated 200 family of proteins that contain a partially conserved 220-amino acid domain. It was shown to promote programmed cell death in several experimental conditions. Researchers found that the expression level of MNDA was critical to respond against DNA damage in embryonic stem cells (Khromov et al., 2012).
As for epigenetic modifications, we compared the co-DE-genes between mRNA expression database and methylation data sets, and identified several Hyper-LGs. Those genes were LILRA2, PLEK, EVI2A, GMFG, ESR1, MMP10, PDLIM4, POU2F2, IL10, and TLR4. It was found that DNA methylation-induced inhibition of some gene expression is part of the reason for normal placental development (Novakovic et al., 2008). Abnormal DNA methylation was found in the decidual chorionic villi of URSA with normal karyotype, especially at the loci of the imprinting genes (Hanna et al., 2013; Zheng et al., 2013; Yu et al., 2018). Thus, the mentioned Hyper-LGs might be important for the occurrence and development of URSA.
The findings of the miRNAs–mRNAs network revealed a high degree of hsa-miR-498 and hsa-miR-4530, indicating that these miRNAs might play key roles in the development of URSA. According to our predictions, hsa-miR-498 negatively regulated BTK, NAMPT, CD86, FCGR1A, LMO3, NCF2, ESR1, IQCH, and MS4A7. hsa-miR-4530 negatively regulated multiple genes, including SLC1A2, ALDOB, SCD, and TENM4. However, little research has been done on these two miRNAs at present.
Conclusion
This study identified several genes and miRNA that involved in the development and progression of URSA, including FCGR1A, FCGR3A, CXCL8, HCK, PLEK, IL10, EVI2A, GMFG, ESR1, MMP10, hsa-miR-498, and hsa-miR-4530. Although further in vivo and in vitro validations are required, our results provide important information to elucidate the pathological process of URSA and may provide a theoretical basis for future studies.
Footnotes
Acknowledgment
This study was supported by grants from Key Research and Development Programs of Science and Technology Bureau of Sichuan Province (Grant No. 2019YFS0421).
Authors' Contribution
H.C. was responsible for searching the GEO database, analyzing the data, and writing the article. S.C. was responsible for analyzing the data using R language. C.L. and J.F. were responsible for enrichment analyses and visualizing the data. W.H. was responsible for editing the article.
Author Disclosure Statement
The authors declare they have no financial conflicts of interest.