
Abstract
This study intended to identify biomarkers for septic cardiomyopathy (SC). Microarray data GSE79962 including 20 SC samples and 11 normal samples were downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) between SC and control groups were identified, followed with functional enrichment analyses. In addition, the protein–protein interaction (PPI) network and modules were constructed. Finally, a transcription factors (TFs)-microRNA (miRNA)-target gene network was constructed and the potential drugs targeting key DEGs were searched. There were 119 upregulated and 80 downregulated genes in the SC group compared with the control group. The upregulated DEGs were significantly enriched tumor necrosis factor signaling pathway, Jak-signal transducer and activator of transcription (STAT) signaling pathway, hypoxia-inducible transcription factor-1 signaling pathway, chemokine signaling pathway, and cytokine–cytokine receptor interaction. The downregulated genes involved in biological processes of negative regulation of DNA biosynthetic process, and skeletal muscle cell differentiation. CCL2, STAT3, MYC, and SERPINE1 were hub nodes in the PPI network and modules. miR-29 family and miR-30 family were considered as key miRNAs, and TATA, MEF2, and STAT5B were considered as key TFs. SERPINE1 and MYC were also drug target genes. The identified DEGs and pathways may be implicated in the progression of human SC, which may lead to a better understanding of SC pathogenesis.
1. Introduction
Sepsis results in millions of deaths worldwide annually (Deutschman and Tracey, 2014). In China, >4 in 1000 people suffer from sepsis, and at least 3.51 million cases with severe sepsis need to be treated per year (Liao et al., 2016). End-organ damage in sepsis has influences on the significant organs of the body, including heart. Septic cardiomyopathy (SC) is one of complications of severe sepsis and septic shock (Beesley et al., 2018; Court et al., 2002). Earlier treatments recommended guidelines include antibiotics treatment, fluid infusion, and vasopressor treatment, which are critical for the outcomes of patients. Currently, there is no available treatment for the specific organ damage in sepsis. The recommended therapeutic strategies for sepsis are symptomatic, except for neutralizing the pathogen with antibiotics. Therefore, the potential molecular mechanisms contributing to the pathogenesis of SC remain to be investigated to screen potential targets for the treatment of SC.
Up to now, many mechanisms have been proposed for implication in SC, including nitric oxide, toxins, cytokines, apoptosis, complement activation, and energy metabolic disorders (Romero-Bermejo et al., 2011; Rudiger and Singer, 2007). Recent studies have shown that genetic disposition was the determinant of cardiomyopathy risk (Jacoby and McKenna, 2012; Leger et al., 2016; Mestroni et al., 2014). However, few studies have focused on the gene expression profiling of human SC sample to uncover the possible mechanisms. Recently, Matkovich et al. (2017) compared the gene expression levels in hearts from patients who died from sepsis and patients with advanced heart failure, who were undergoing cardiac transplantation. They found that there were distinct changes in gene expression in hearts from patients with SC compared with that in patients with end-stage heart failure, which suggested the critical role of genetic factors in SC. Despite numerous differentially expressed genes (DEGs) identified by Matkovich et al. (2017), they did not carried out further analysis to select more key markers as well as to explore the potential regulatory mechanism. Therefore, further analysis using this gene profiling data set may be necessary to unveil novel mechanisms.
In this study, we downloaded the gene expression profile (GSE79962) deposited by Matkovich et al. (2017) from Gene Expression Omnibus (GEO) databases to further uncover the biomarkers associated with SC development and progression. The DEGs between SC samples and controls were identified. Subsequently, the functional enrichment analyses, protein–protein interaction (PPI) network analysis, and transcription factor (TF)-microRNA (miRNA)-target regulatory network analysis were performed for these DEGs. In addition, potential drugs targeting key DEGs were also predicted. We aimed to identify key genes associated with SC as well as further elucidate the potential molecular mechanisms through bioinformatics analysis.
2. Methods
2.1. Data collection
The gene expression profile GSE79962 was downloaded from GEO (Barrett et al., 2007), which was based on the platform of Affymetrix Human Gene 1.0 ST Array (Thermo Fisher Scientific, Inc., Waltham, MA). A total of 31 heart tissue samples in this data set, including 11 from normal heart (control group) and 20 from SC patients, were analyzed.
2.2. Data preprocessing
The robust multichip average integrative algorithm (Bolstad et al., 2003; Irizarry et al., 2003a,b) in Affy package (Gautier et al., 2004; version 1.56.0, http://bioconductor.org/packages/release/bioc/html/affy.html) of R (version 3.4.3) was used for preprocessing the microarray data, including background correction, quartile data normalization, and probe summarization. After this, the probe ID was mapped to gene symbol on the basis of the annotation file on the platform. The probe without gene symbol was deleted. For multiple probes mapped to the same gene, the mean value of these probes was taken as the final expression of this gene. Finally, 22,965 genes out of 323,321 probes were retrieved.
2.3. Identification of DEGs
The limma (version 3.34.9) package in R version 3.4.3 (Phipson et al., 2016; Ritchie et al., 2015) was used to screen out the DEGs between SC and control. p Values were corrected by the Benjamini and Hochberg method, obtaining adjusted p value. Genes with |log fold change (FC)| > 1 and adjusted p < 0.05 were selected as DEGs. Then, cluster analysis was performed using the Pheatmap package (https://cran.r-project.org/package=pheatmap) in R to further analyze the DEGs.
2.4. Enrichment analysis
The Gene Ontology (GO) database consists of three categories, including biological process, molecular function, and cellular component. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways database is a comprehensive and recognized database encompassing numerous biochemical pathways (Kanehisa and Goto, 2000). To gain further insight into the potential functions of the identified DEGs, the Database for Annotation, Visualization and Integrated Discovery (version 6.8) tool (Huang da et al., 2009) was used to perform GO and KEGG pathway enrichment analyses. The thresholds were set as p < 0.05 and enrichment counts >2.
2.5. PPI network analysis of DEGs
The Search Tool for the Retrieval of Interacting Genes (STRING, version 10.0, www.string-db.org) provides comprehensive and easily accessible interaction information derived from known and predicted protein data (Szklarczyk et al., 2017). The PPI between DEGs was predicted from STRING, and the interactions with a combined score of >0.4 were selected. The PPI network was visualized using Cytoscape (version 3.6.1, www.cytoscape.org) software (Shannon et al., 2003). Nodes with higher degree centrality (an indicator of topological property) were considered as hub proteins in the PPI network and may have an essential role in the network.
2.6. Network module analysis
Molecular Complex Detection (MCODE, version 1.4.2, http://apps.cytoscape.org/apps/MCODE) could detect densely connected regions in the PPI network (Bader and Hogue, 2003). At present, the clusters of the highly intraconnected nodes with the following thresholds degree cutoff = 2, node score cutoff = 0.2, k-core = 2, maximum depth = 100, and score ≥5 were screened from the PPI network using MCODE.
2.7. TF-miRNA-target gene regulatory relations analysis
The overrepresentation enrichment analysis method in Webgestalt (Wang et al., 2013; Zhang et al., 2005; http://www.webgestalt.org) tool was used to predict the TF and miRNAs of DEGs. The threshold was designed as p value <0.05. Based on the predicted TF-target gene and miRNA-target gene pairs, the TF-miRNA-target gene regulatory network was visualized using Cytoscape.
2.8. Prediction of drugs targeting DEGs
The Drug–Gene Interaction Database (DGIdb, www.dgidb.org) is a web resource that organizes and presents gene druggability information and drug–gene interactions from databases, articles, and web resources (Cotto et al., 2018). In this study, the potential drugs targeting key DEGs confirmed by network module analysis were predicted using DGIdb (version 3.0.2) with the following parameters: preset filter, Food and Drug Administration approved; advanced filters, source databases: all; gene categories, all; and interaction types, all. The interaction network was constructed by Cytoscape.
3. Results
3.1. Identification of the DEGs
A total of 199 DEGs were obtained between SC and control groups, including 119 upregulated genes and 80 downregulated genes. As shown in the heat map (Fig. 1), the DEGs were predominantly separated into the clusters: SC versus control.

Heatmap results of DEGs. DEGs, differentially expressed genes.
3.2. Function and pathway enrichment analyses
The upregulated DEGs were significantly enriched in inflammatory response associated biological process (BP) terms (Fig. 2a), whereas downregulated DEGs were enriched in skeletal muscle cell differentiation (Fig. 2c). Pathway analysis showed that upregulated DEGs were enriched in malaria, hypoxia-inducible transcription factor-1 (HIF-1) signaling pathway, cytokine–cytokine receptor interaction, Jak-signal transducer and activator of transcription (STAT) signaling pathway, chemokine signaling pathway, and tumor necrosis factor (TNF) signaling pathway (Fig. 2b), whereas downregulated DEGs were significantly enriched in miRNA in cancer (Fig. 2d).
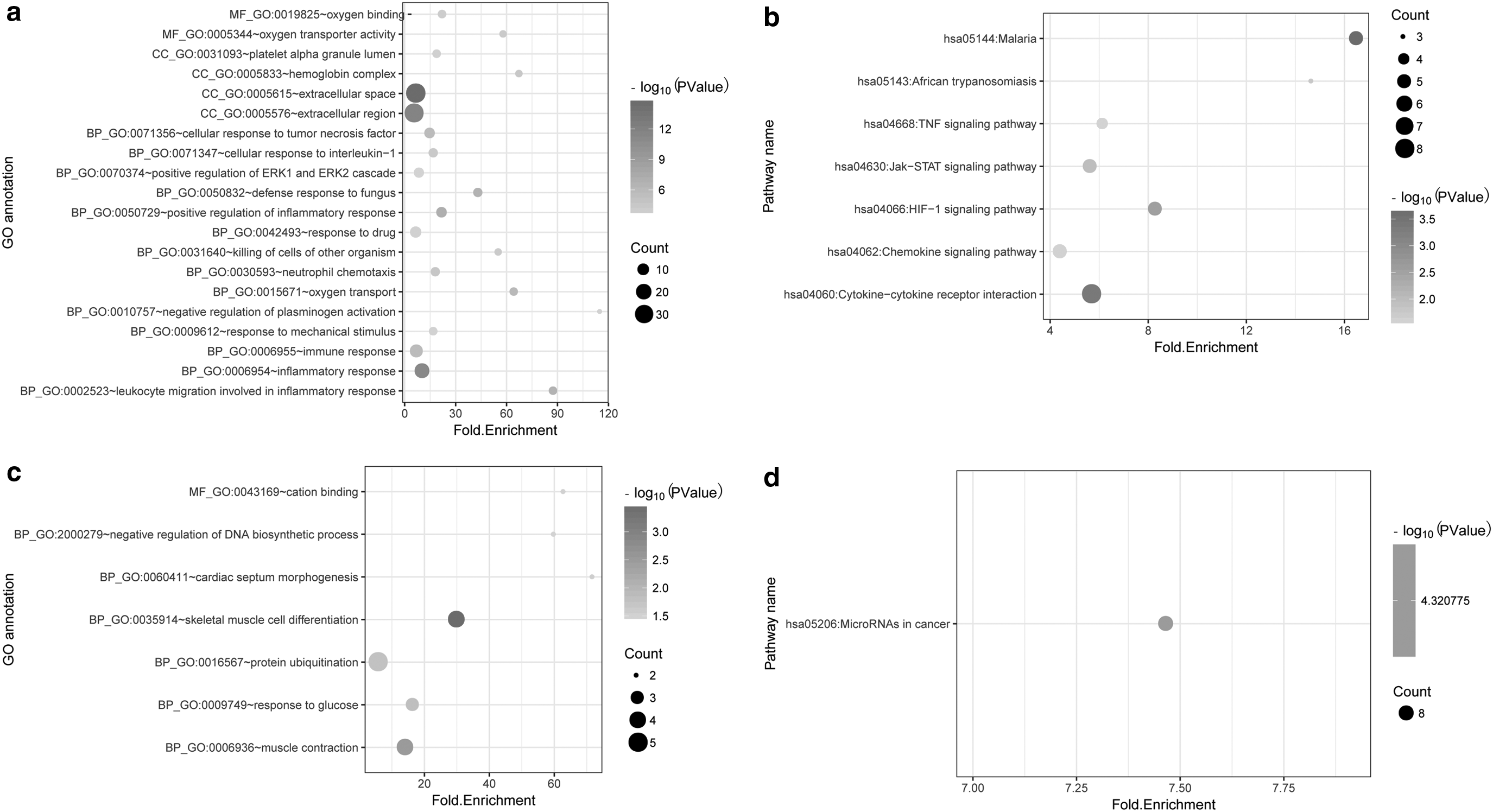
The DEGs significantly enriched GO (top 20) and Kyoto Encyclopedia of Genes and Genomes pathways.
3.3. PPI network analysis of DEGs
In the PPI network, there were 92 nodes (proteins) and 186 edges (interactions; Fig. 3a). The 92 nodes included 67 upregulated genes and 25 downregulated genes. According to the topology score, C–C motif chemokine ligand 2 (CCL2), STAT3, and MYC proto-oncogene, BHLH TF (MYC), had the top three high degrees (Table 1). In addition, one submodule with score >5 was extracted from the PPI network, which included 16 nodes and 42 edges (Fig. 3b). In this module, CCL2, MYC, and serpin family E member 1 (SERPINE1) had the top three high degrees (Table 1).
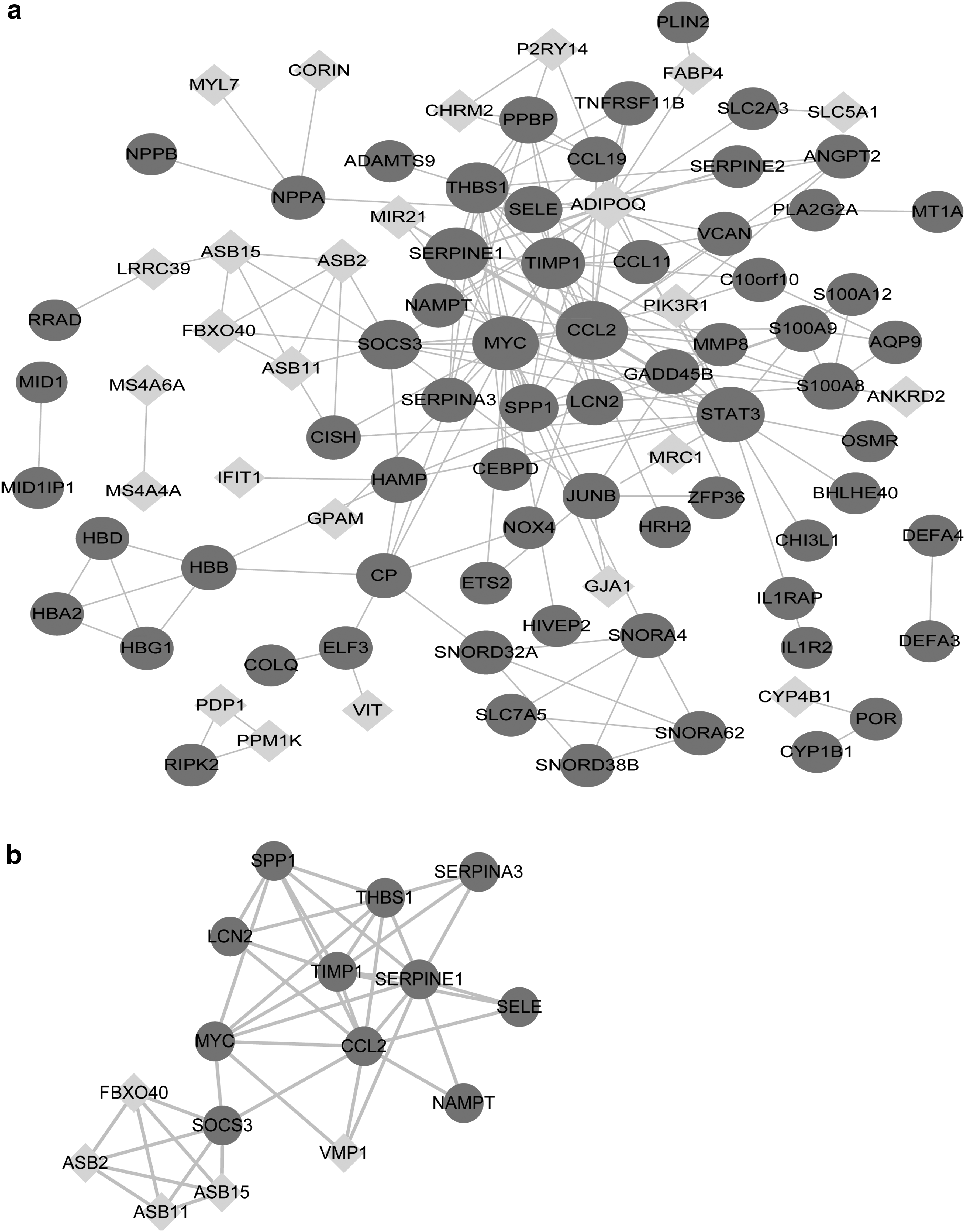
The protein–protein interaction network
Top 10 Nodes with Higher Degrees in Protein–Protein Interaction Network and Module
Degree: topological degree score.
3.4. TF-miRNA-gene network analysis
The enriched miRNA and TFs from DEGs are given in Tables 2 and 3, respectively. miR-29 family and miR-30 family regulated the most target genes. Among TFs, TATA-box binding protein (TATA), myocyte enhancer factor 2A (MEF2), and STAT5B regulated more targets. In the constructed TF-miRNA-gene regulatory network, a total of 96 nodes and 209 interactions were included, as shown in Figure 4.
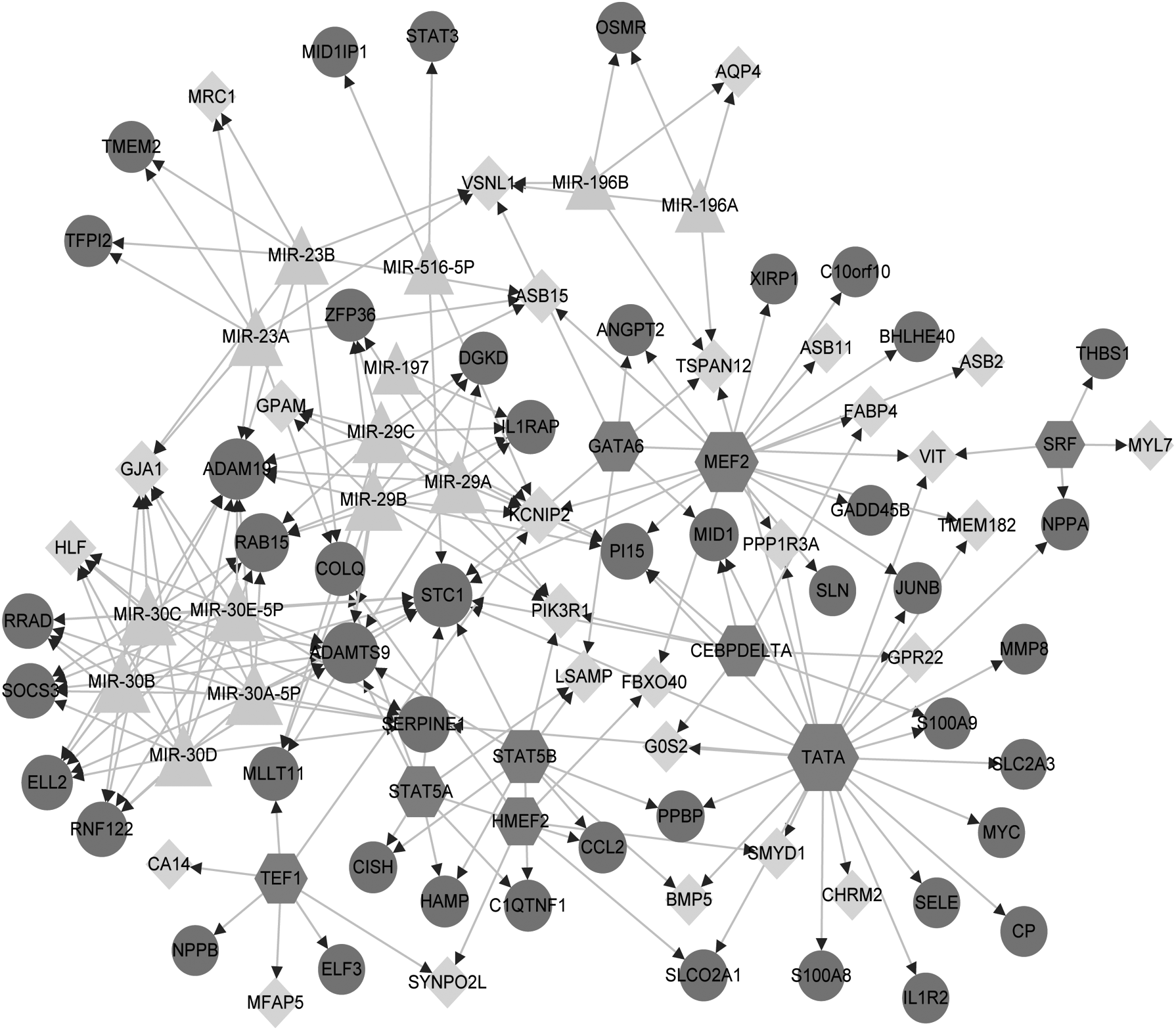
The constructed TF-miRNA-target gene network. The dark grey circle represents the upregulated gene, the light grey prism represents the downregulated gene, the light grey triangle represents the miRNA, and the dark grey hexagon represents the TF. miRNA, microRNA; TF, transcription factor.
Regulatory MicroRNAs of Differentially Expressed Genes Screening
C, gene count of target sequence; E, expectation value; Geneset, target sequence and microRNAs; O, gene number provided by users; overlap gene user ID, gene list of target gene of related microRNAs; p value: p value after hypergeometric test; R, enrichment ratio.
Regulatory Transcription Factors Screening
Overlap gene user ID, gene list of target gene of related transcription factors.
3.5. Drug prediction
The 16 nodes in the obtained module (score >5) were considered key genes and were further analyzed by DGIdb. Finally, seven genes were considered as druggable genes, such as SERPINA2, SERPINE1, and MYC. Based on the seven druggable genes, an interaction network including 81 nodes and 75 edges was constructed, as shown in Figure 5.
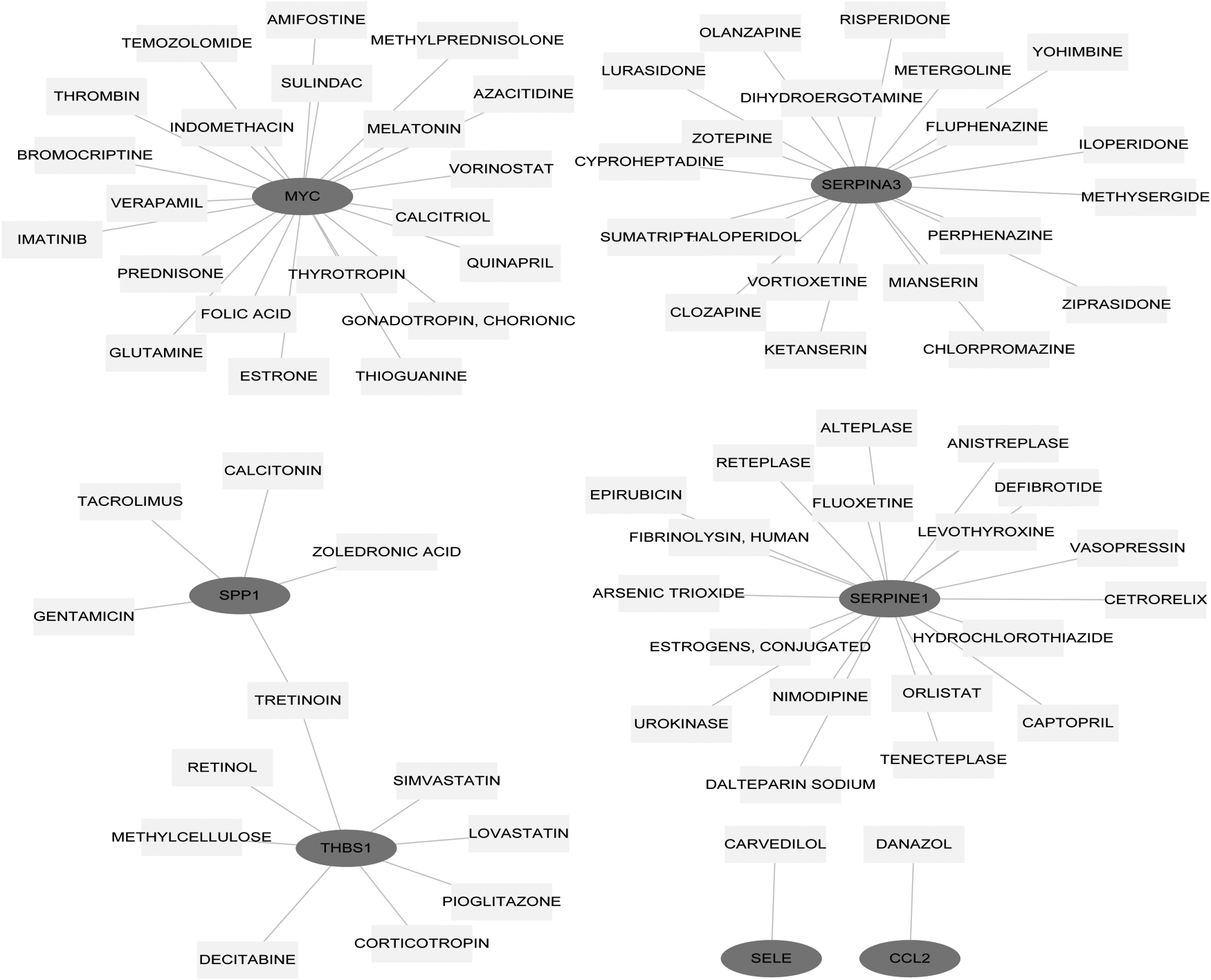
The constructed drug target gene interaction network. The dark grey circle represents the upregulated gene and the light grey rectangle represents the drug.
4. Discussion
This study identified 199 DEGs between SC and control groups. Function analyses showed that the upregulated genes were enriched TNF signaling pathway, Jak-STAT signaling pathway, HIF-1 signaling pathway, chemokine signaling pathway, and cytokine–cytokine receptor interaction. The downregulated genes were associated with biological processes of negative regulation of DNA biosynthetic process, and skeletal muscle cell differentiation. Moreover, CCL2, STAT3, MYC, and SERPINE1 were identified as hub nodes in the PPI network and modules. miR-29 family and miR-30 family were considered as key miRNAs, and TATA, MEF2, and STAT5B were considered as key TFs.
CCL2 had the highest degree in the PPI network and module 1. Besides, it significantly enriched in chemokine signaling pathway, TNF signaling pathway, and cytokine–cytokine receptor interaction. CCL2 is a cytokine involved in immunoregulatory and inflammatory processes (Hubal et al., 2010). Recent studies have reported an increased expression level of CCL2 in a lipopolysaccharide-induced inflammatory model using human-induced pluripotent stem cell-derived cardiomyocytes (Yucel et al., 2017). The three pathways mentioned are all inflammation-associated pathways (Bradley, 2010; Keane and Strieter, 2000; O'Shea and Murray, 2008). A recent study has reported that excessive production of proinflammatory mediators plays a key role in the process of sepsis-induced myocardial dysfunction (Moussa et al., 2015). Therefore, we speculated that CCL2 may involve in the SC pathogenesis through these inflammatory pathways.
STAT3 is a TF for numerous proinflammatory genes expression (Bollrath and Greten, 2009). Activated STAT3 acts as an important TF to regulate interleukin (IL)-17 expression (Camporeale and Poli, 2012). Increased IL-17 production through increased STAT3 transcription by CD4+ T cells has been indicated as an indicator of postseptic immune dysfunction (Mukherjee et al., 2012). In this study, STAT3 was also one of the enriched genes in HIF-1 signaling pathway. HIF-1 regulates numerous genes that control cellular processes essential to the cardiovascular system (Shohet and Garcia, 2007). Most recently, He et al. (2018) reported that edaravone could improve septic cardiac function by activating the HIF-1α/heme oxygenase-1 pathway, which suggested the important role of HIF-1 signaling pathway in SC. Therefore, STAT3 may play a role in SC development through HIF-1 signaling pathway.
In addition, MYC encodes a nuclear phosphoprotein that participates in processes of apoptosis, cell cycle, and cellular transformation (Kalkat et al., 2017). It has been reported that cell death and apoptosis play a key role in the progression of SC (Tsolaki et al., 2017). Besides, MYC was involved in Jak-STAT signaling pathway that has been shown to be associated with the release of inflammatory mediators and cytokines and involved in the regulation of immune response in sepsis (Cai et al., 2015). Specially, MYC was one of regulated genes of TF TATA. SERPINE1, also known as PAI-1, is expressed in cardiomyocytes, the upregulation of which could result in the development of interstitial and perivascular fibrosis in the postmyocardial infarction heart (Takeshita et al., 2004). Moreover, increased level of SERPINE1 was also reported in sepsis (Zeerleder et al., 2006). SERPINE1 was also involved in HIF-1 signaling pathway. Interestingly, SERPINE1 and MYC were predicted to be druggable genes of SC. Taken together, MYC and SERPINE1 as well as TF of TATA may serve as potential biomarkers of SC.
The miR-29 family consists of miR-29a, miR-29b, and miR-29c (Kriegel et al., 2012). It has been reported that the areas bordering the infarcted myocardium present a reduced expression of the three miR-29 isoforms in mice (van Rooij et al., 2008). Cardiac pressure overloading has been shown to decrease the expression of miR-29c in animal models (van Rooij et al., 2008). miR-30 family is highly expressed in cardiomyocytes (Zhu et al., 2009). It is also found to regulate angiotensin-II-induced myocardial hypertrophy through regulating autophagy (Pan et al., 2013). Downregulation of miR-30c could enhance myocardial matrix remodeling in pathological left ventricular hypertrophy (Duisters et al., 2009). Given the role of miR-29 and miR-30 families in myocardial function, we speculated that they may play key roles in SC progression.
In summary, our study identified some key genes, pathways, and drug target genes closely associated with SC using bioinformatics analyses. The key genes such as CCL2, STAT3, MYC, and SERPINE1 might serve as potential biomarkers and therapeutic targets in SC. These identified pathways may provide a molecular mechanism underlying SC progression.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.