
Abstract
The folding of a protein structure is a process governed by both local and nonlocal interactions. While incorporating local dependencies into a machine learning algorithm for protein structure prediction is simple and has been exploited for some time, the modeling of long-range dependences which result from structurally-neighboring residues has only recently begun to be addressed. Structural properties designed to localize the prediction space from direct tertiary structure prediction, such as secondary structure, contact maps, and intrinsic disorder, among others, have begun to greatly benefit from machine learning models capable of modeling a widened, potentially global protein context. This has led to a direct enhancement of the quality of predicted tertiary structures through both the optimization of structural constraints and improved reliability of alignments to structural templates. These improvements have stemmed from the application of recurrent and convolutional neural network architectures effective not only at innate sequential context propagation but also deep feature extraction due to novel skip connections and normalization techniques allowing for greatly enhanced error back-propagation. The recent results from independent blind testing in Critical Assessment of protein Structure Prediction 13 have signaled the beginning of a new generation of protein structure prediction through the utilization of these contextual techniques. The ripples from advancements in the determination of one-dimensional and two-dimensional structural properties have us moving ever closer to the solution of the protein structure prediction problem.
1. INTRODUCTION
Proteins are biological macromolecules consisting of chains of evolution-derived combinations of 20 standard amino acids linked together with peptide bonds. They perform a myriad of crucial biological roles across all domains of life. Such diverse functional roles commonly rely on a wide variety of unique tertiary [i.e., three-dimensional (3D)] structures (shapes) for different proteins (Nelson et al., 2008). Experimental determination of a protein structure, obtained through methods such as X-ray crystallography, cryogenic electron microscopy, and nuclear magnetic resonance (NMR) spectroscopy (Wuthrich, 1989; Drenth, 2007; Frank, 2017), is a slow and expensive process, illustrated by the growing gulf between known protein sequences deposited in the UniProt database and solved structures in the Protein DataBank (Berman et al., 2006; UniProt Consortium, 2018). Moreover, not all structures are suitable for structure determination due to limitations of various experimental techniques. Thus, solving or predicting protein structures computationally is the only viable solution for millions of proteins with unknown structure.
The foundation of protein structure prediction was laid by Anfinsen (1973) in his groundbreaking work which postulated that the tertiary structure of a protein is wholly determined by its amino acid sequence (or primary structure) alone. Since then, modeling this folded structure through computational algorithms has been a key focus in structural bioinformatics. The deterministic nature of the folding path undertaken by a denatured protein is a tenet of Levinthal's paradox, which states that a protein cannot possibly arrive at its native conformation by way of random sampling of the conformational plane, due to the multitude of possible structural conformations (estimated to be around 10300 possible structures) and relatively quick folding process (roughly 1 second from random coil to folded state; Levinthal, 1969). While this process can theoretically be interpreted by an energy landscape model which minimizes the free energy of the resultant folded structure, there currently is no optimal energy function available (Zhou et al., 2011).
Due to these immense complexities, protein 3D structure prediction is often broken down into simpler one-dimensional (1D) and two-dimensional (2D) structural descriptors. These schemas are designed to not only provide a low-level descriptor of the folded 3D structure (Murzin et al., 1995) but also to provide insights on a protein's functionality and/or evolution in their own right (Zhou and Zhou, 2005; Godzik et al., 2007). In the end, it is hoped that the solution to these subproblems will culminate in the solution of the overall protein structure prediction problem, as demonstrated by recent progress in the Critical Assessment of protein Structure Prediction techniques (CASP; Moult et al., 2018). In this article, we use the term “protein structure prediction” to refer to the prediction of both tertiary structure and any of the structural descriptors.
The aim of this review is to discuss the current status of the hierarchy of protein structure prediction, with a perspective from contextual neural network architectures. The previous review from Min et al. (2017) provided a thorough overview on the role of deep learning in the larger bioinformatics fields circa 2017. Litjens et al. (2017), Mamoshina et al. (2016), and Libbrecht and Noble (2015) have performed similar reviews in other omics fields (medical imaging, biomedicine, and genetics/genomics, respectively). Other reviews, such as those by Huang et al. (2016), Necci et al. (2018), Liu et al. (2017), Jiang et al. (2017), Yang et al. (2018), and Chen et al. (2018b), have provided recent reviews on the current status of specific protein prediction subproblems. Paliwal et al. (2015) wrote a very brief, but similar analysis on deep learning applications in protein structure prediction. Another similar review based on early machine learning models was written by Cheng et al. (2008). In this study, we will focus on contextual machine learning in structural bioinformatics.
2. CONTEXTUAL MACHINE LEARNING
2.1. A basis for sequence learning
Sequence learning is inherent to human psychology in our ability to process simple sequential events to understand their cause and effect. For example, when reading a book, humans do not process each letter or word separately to understand its pragmatics, but innately build a gradual and adaptive understanding through fragments, sentences, and paragraphs of related words. This buildup of context allows us to make more educated decisions about not only the information provided but also can be used to estimate future or unknown events due to the association of the surrounding context and the locality of the unknown entity (Spiegel and McLaren, 2006).
While this ability is natural to human learning, machine learning approaches must be adapted to incorporate these contextual patterns into their predictions. Encoding the dependencies throughout sequential data points is the key to solving numerous problems, particularly sequence-dependent speech, image, and natural language processing tasks, which have been major hubs of innovation in this technology (Lipton et al., 2015). The dependencies present in these problems can be local ideas conveyed by adjacent data points and nonlocal longer-range dependencies spread throughout a sequence. The length of these dependencies and the context needed to accurately represent sequential patterns are problem dependent; natural language processing of singular sentences will naturally need less context than audio processing of a signal sampled at 8 kHz. One key aspect to the modeling of intersequence dependencies is therefore the efficiency of propagation throughout the sequence so that information is preserved throughout its appropriate range (Hochreiter et al., 2001).
Several machine learning architectures have proved capable of sequence learning throughout its short history, all achieving various levels of context propagation. These methods, dubbed herein as “contextual machine learning algorithms,” are proving valuable to researchers due to their ability to effectively and efficiently model the surrounding information for the prediction of a singular data point in multidimensional data arrays. The inclusion of context in these models has allowed them to learn from a richer source of information, forming a representation benefiting from the inclusion of underlying location-dependent motifs in the data. Some of these models, discussed in the next section, have only recently become viable methods of prediction due to the rise of available data and accessibility of powerful computational resources.
2.2. Algorithms for modeling context
Early on in sequence analysis, context was forcibly introduced through a sliding window surrounding the input data sample compatible with then-current discriminative applications of shallow artificial neural networks (ANNs; Rumelhart et al., 1986), support vector machines (SVMs; Vapnik, 1998), and random forest classifiers (Breiman, 2001). However, the learning of intrasequence dependencies can be weak due to the large input often being inaccurately modeled by the limited representational power of the hidden layers in the network. Furthermore, the context of the model is limited to the relatively small window (typically N is between 5 and 20 residues), when important long-range dependencies can be interspersed much more deeply throughout the sequence in many applications. An example of a single-layer neural network (NN) with a window size of 2 is shown in Figure 1A.
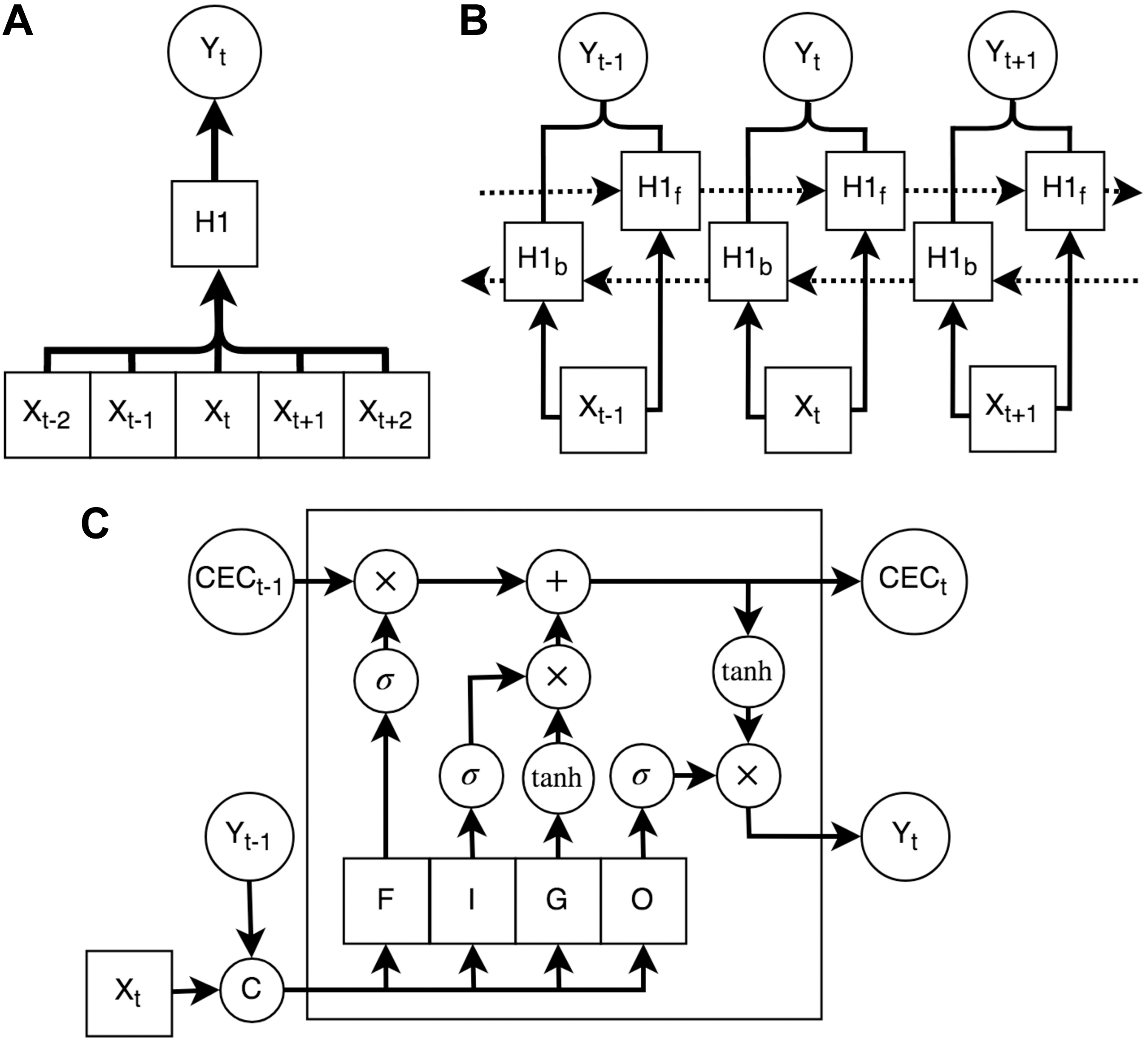
The evolution of dependence propagation in recurrent neural network. The windowing method (for a window size of 2) for prediction of sequence position t through a one-layer neural network is shown in
Recurrent neural networks (RNNs) overcame these weaknesses by encoding the dependencies between adjacent steps in a sequence (Hopfield, 1982; Werbos, 1990). This achieves a theoretical global context window by passing information from one end of the sequence to the other. As many dependencies are not unidirectional, bidirectional recurrent neural networks (BRNNs) allow bidirectional propagation of dependencies by splitting the forward and backward sequential connections (Schuster and Paliwal, 1997), as shown in Figure 1B. A weakness of vanilla RNN architectures is that they empirically lack the ability to effectively model long-term dependencies due to affine transformations vanishing or exploding the error gradients (Hochreiter et al., 2001). Long short-term memory (LSTM) cells attempt to amend this problem by introducing a dedicated memory bus [called the constant error carousel (CEC)] to allow unimpeded error backpropagation through the sequence (Hochreiter and Schmidhuber, 1997). The architecture of a LSTM cell is shown in Figure 1C. Another architecture proposing the same benefits with reduced parameter counts was proposed in Cho et al. (2014).
Convolutional neural networks (CNNs; LeCun et al., 1989) are another contextual machine learning model, in that they utilize a widened receptive field through multidimensional weights to gradually increase their contextual window through the layers of the network. This is similar to windowed ANNs (a one layer CNN is in fact a windowed ANN), but the context aggregates through the network rather than remaining fixed. A strength of CNN is their generality due to their lower parameter counts and their ability to learn local spatial/sequential coherence. As is shown in Figure 2, the depth of a CNN and the width of its convolutional kernel are the deciding factors in how far through the sequence the context window can reach. Thus to attain global context in proteins, the kernel size or number of layers must be significantly high, particularly for long proteins.
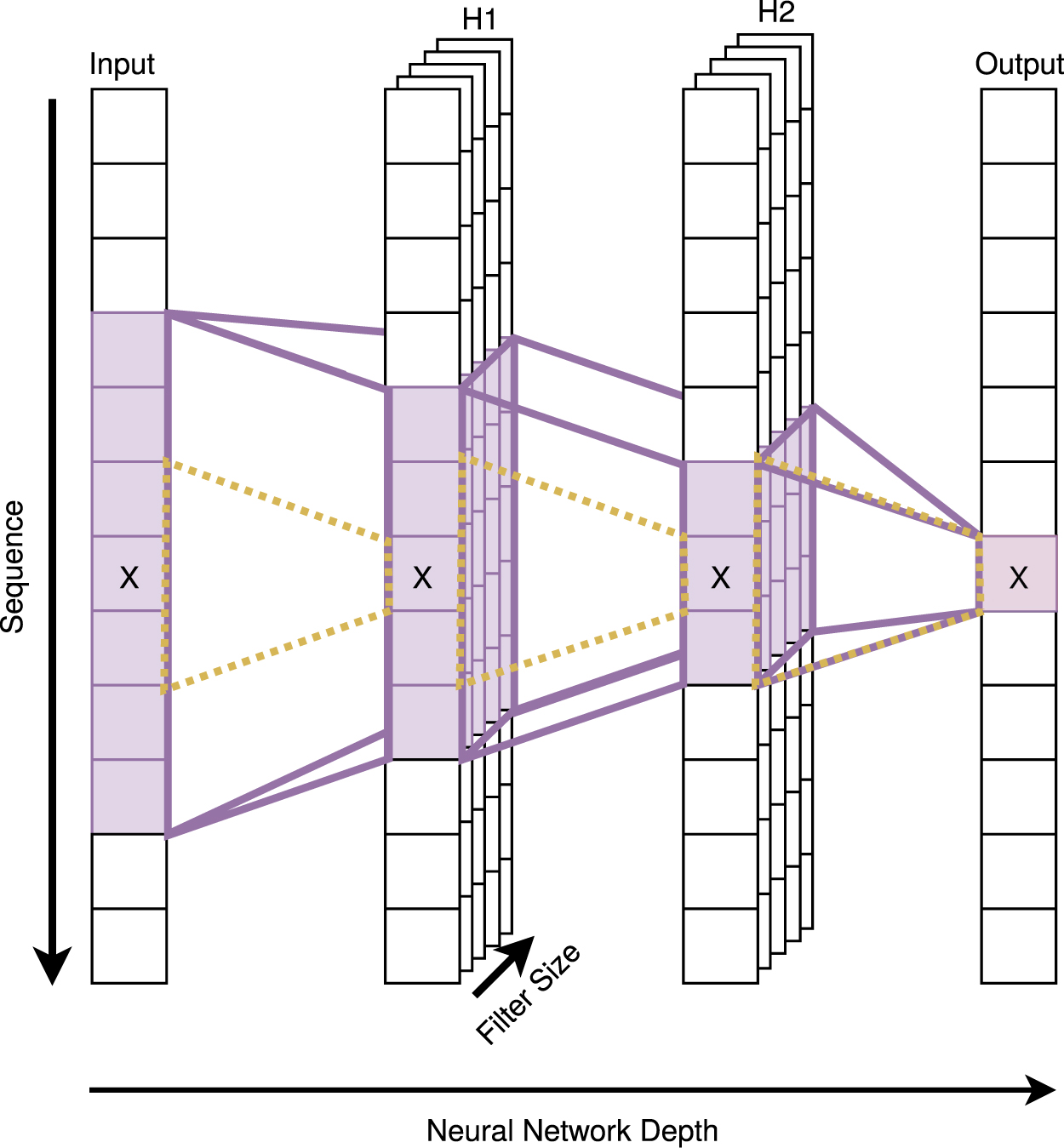
An illustration of how context expands with the number of layers in a one-dimensional convolutional neural network with two hidden layers (H1 and H2) for sequence position “x.” The dashed yellow cone represents the context immediately available at position x from layer to layer.
As smaller kernel sizes are generally preferred for increased feature abstraction and parameter minimization, the onus of increasing the contextual window has generally been on the depth of the network. Increasing the depth of a vanilla CNN architecture is problematic due to the vanishing gradient problem (Bengio et al., 1994), but the innate compromise between effective gradient propagation and context exposure was broken by the introduction of skip connections in ResNets (He et al., 2016a,b) and highway networks (Srivastava et al., 2015), in which the backpropagated error signal can easily travel back through dedicated paths in the network. The application is akin to the use of the CEC in LSTM cells, but aids error propagation through the layers in the network rather than through the sequence. More recent developments in this field include the incorporation of multiple “inception”-style convolutional paths into the residual block (Szegedy et al., 2017), decision gates for each residual block in squeeze and excitation networks (Hu et al., 2018b), grouping multiple convolutions in the residual blocks (Xie et al., 2017), and the application of residual connections in LSTM layers (Kim et al., 2017).
2.3. Application to proteins
Similar to human language, each protein can be thought of as a sequence of “words” constructed with an alphabet of 20 base characters (amino acids). Just as different combinations of words in paragraphs lead to different semantic meanings representative of both content and context, different sequences of amino acids lead to different structures with a directly corresponding 3D representation. However, unlike sentence structure, key interactions in proteins induced by structurally-neighboring amino acid residues may be separated by hundreds of residues. Detecting these nonlocal but structurally-neighboring interactions provides key information toward the solution of the protein structure prediction problem. The lack of the ability to capture these nonlocal effects has been a long-term obstacle for solving the problem even for 1D structural properties such as secondary structure (SS) and local backbone angles (Jiang et al., 2017; Min et al., 2017; Yang et al., 2018). However, the recent advances in contextual machine learning resulting from audio and image works described above have recently begun to bear fruits in protein structure prediction in 1D, 2D, and finally 3D, defining new levels of performance sweeping through the literature.
The rapid increase of protein structure prediction publications utilizing these contextual techniques is shown in Figure 3. These data are obtained by searching Web of Science in April 2019 for articles containing “protein” and “structure” and “recurrent neural*” or “convolutional” in their topical information. This graph shows that the past 2 years have accounted for 52% of all articles published in the 27-year search, with 7 more already published as of April 2019.
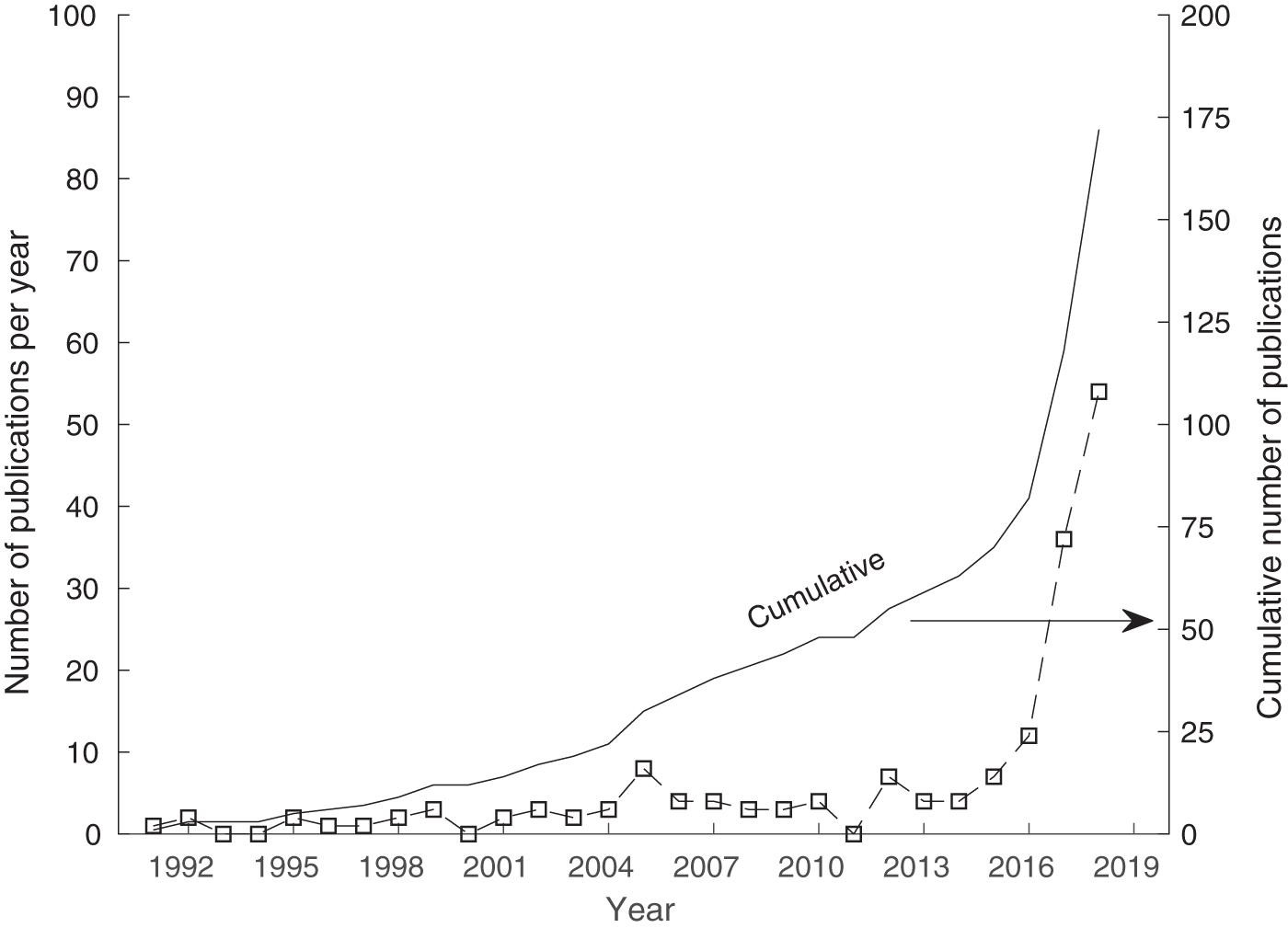
The rise of contextual machine learning publications in structural bioinformatics, obtained from the Web of Science.
3. PREDICTION OF 1D STRUCTURAL PROPERTIES
3.1. Protein secondary structure
Perhaps the most well-known structural property of proteins is that of protein secondary structure. Secondary structure is a 1D representation of the local protein backbone through categorization of the local structure into several template classes. This representation can categorize the structure of a protein into protein folds classified by the layout of a protein's secondary structure elements (Murzin et al., 1995; Dai and Zhou, 2011). Protein secondary structure also provides insight into the folding path undertaken by a protein (Zhou and Karplus, 1999). For a more comprehensive review on the history of the secondary structure prediction problem, see the reviews by Yang et al. (2018), Jiang et al. (2017), and Pirovano and Heringa (2010).
First described by Pauling et al. (1951) in their analysis of helical and sheet hydrogen bonding patterns in a protein backbone, protein secondary structure has been at the forefront of structural bioinformatics for decades. This is because the packing of secondary structure elements directly impacts the folding path of a protein, a process governed by nonlocal interactions. Many early models utilized a sliding window of typically <25 amino acid residues (Faraggi et al., 2012; Yaseen and Li, 2014; Heffernan et al., 2015; Spencer et al., 2015). DeepCNF had previously utilized a CNN-based architecture with a posteriori conditional modeling on the outputs (Wang et al., 2016b), but the small size of the network limited the contextual exposure. A BRNN method was first used in Porter4 (Mirabello and Pollastri, 2013), which used a vanilla RNN architecture. The long-range contextual modeling of Porter4 was outperformed by SPIDER3, a LSTM-BRNN-based iterative model which was the first attempt to efficiently use whole-sequence learning (Heffernan et al., 2017). SPIDER3 managed to improve on its predecessor SPIDER2 by >2.5% in SS3 accuracy despite it using the same input features and training data. The method was particularly more effective for residues with a high number of nonlocally interacting residues (i.e., long-range interactions; Heffernan et al., 2017).
The development of this method spurred a new generation of contextual learning algorithms. Many of these methods have turned to deeper architectures to enhance the feature abstraction power of the networks to improve on SPIDER3. This includes methods based on Inception-DenseNets in MUFOLD-SS (Fang et al., 2018), an LSTM-BRNN and wide CNN in NetSurfP-2.0 (Klausen et al., 2019), and an ensemble of LSTM-BRNN/ResNets in Porter5 and SPOT-1D (Hanson et al., 2018b; Torrisi et al., 2018). The latest method, SPOT-1D, has further improved on SPIDER3 by >2.3% in SS3 prediction to reach 86% accuracy, while simultaneously achieving 75% in SS8 prediction, indicating that some of the faint long-range dependencies governing the folding of secondary structure only become detectable as the network becomes sufficiently deep. The accuracies reported by these methods are shown in Table 1, sorted by their publication date.
A Comparison of the Reported Accuracies by Recently Released Contextual Secondary Structure Predictors and the Datasets the Results Were Obtained From
3.2. Protein backbone angles
As secondary structure is an inherently coarse-grained descriptor, this representation is often complemented by real-valued angles of the torsion and dihedrals along the protein backbone. These angles can be used to sequentially construct a general model tertiary structure of a protein chain at a much reduced prediction space than direct prediction of the Cartesian coordinates (Parsons et al., 2005), with the additional benefit of rotational or translational invariance. Torsion angles φ, ψ, and ω describe the rotation of the plane about the C α –N, C α –C, and C–N bond through the peptide bonds, respectively (Ramachandran et al., 1962). The prediction of angles φ and ψ was initially focused on the classification of the angle into discrete angle bins (Kang et al., 1993; Kuang et al., 2004), but have since also been predicted as a continuous value to provide a more precise estimation (Wood and Hirst, 2005; Heffernan et al., 2015, 2017, 2018; Fang et al., 2019; Hanson et al., 2018b; Klausen et al., 2019). Modeling the angles of a protein backbone requires that the long-range dependencies be maintained to avoid generating unlikely protein conformations or steric clashing in distant sequence positions. This is evidenced by the drop in mean absolute error (MAE) between the window-based method SPIDER2 (Heffernan et al., 2015) and the latest whole-sequence-based method SPOT-1D (Hanson et al., 2018b) of 20.7° to 16.9° for φ and 34.6° to 24.9° for ψ, respectively.
It should be noted that while continuous prediction offers a more exact measurement of the angle under analysis, it offers no information on the confidence or distribution of angles at that position. This distribution can offer indirect insight into other aspects of protein structure, such as protein intrinsic disorder, while still offering an accurate angle measurement depending on the bin size (Gao et al., 2016, 2018). To this end, Xu (2018) has predicted a real-valued discrete distribution of protein torsion at each residue.
Opposed to the continuous-valued prediction of φ and ψ, the ω angle is generally restricted to two isomeric states, namely the cis (0°) and trans (180°) isomers. Since over 99.6% of residues' ω angles are sterically restrained to the trans-isomer (Singh et al., 2018), this class is often assumed and the angle ignored in angle prediction. However, this propensity increases to 4.9% for Proline residues, as Proline is the only amino acid with a side chain regularly permitting the formation of the cis-isomer that is stabilized by nonlocal interactions. As a result of this class imbalance, many of the ω angle classification works have been focused solely on Proline residue prediction (Song et al., 2006; Exarchos et al., 2009a; Yoo et al., 2014; Al-Jarrah et al., 2015) and/or were trained and tested on manually-balanced datasets incompatible with real-world examples (Pahlke et al., 2004; Exarchos et al., 2009b). A recent predictor was trained using an ensemble of ResNet and LSTM models (Singh et al., 2018) to model the long-range sparsity of cis-isomers, to be able to detect the few positions (if any) in a protein sequence energetically favorable for a cis-isomer at 24% sensitivity and 50% precision for proline. However, low sensitivity rates (at defined precision limits) illustrate the difficulty of modeling such a sparse pattern.
There is also another set of angles that describe the position of the C
α
atoms across multiple residues. Angles θ and τ represent the angle formed by
3.3. Protein disorder prediction
The previous structural properties have all been derived from the underlying dogma of the structure–function paradigm, in that a protein's static folded structure provides its innate function. This has been complemented by an understanding of unstructured regions in the form of Intrinsically Disordered Regions and Intrinsically Disordered Proteins, which do not possess a rigid structure in their native physiological conditions (Wright and Dyson, 1999). These proteins often possess an ensemble of transient interconverting structures (Uversky, 2016), providing them with unique characteristics which make them particularly suitable for specialized roles in regulatory functions, cell signaling, and molecular recognition and assembly (Dyson and Wright, 2005; Oldfield and Dunker, 2014). Intrinsic disorder is abundant in all domains of life, but has a particular prevalence in eukaryotes (19.6% of residues) and viruses (9.6% of residues; Hu et al., 2018a), illustrating the impact that the existence of a disorder has on the determination of the larger overall structure (or lack of structure) of a protein.
A difficulty in modeling intrinsic disorder in protein is its varying characterization, categorizing disorder into several “flavors” depending on experimental factors and attributes of the disordered sequence elements (Vucetic et al., 2003; Necci et al., 2018). One example of a disorder flavor is the length of the disordered region, where experimental analysis of disorder has found long-range disordered regions to differ from short-range regions through their propensities for certain amino acid compositions (Tompa, 2002; He et al., 2009). Earlier disorder prediction methods often found it beneficial to train on individual flavors and either release an individual model combining the separate definitions' models (Xue et al., 2010; Zhang et al., 2012) or to release them independently for flavor-specific prediction (Romero et al., 2001; Linding et al., 2003a; Dosztányi et al., 2005; Hirose et al., 2007; Shimizu et al., 2007; Walsh et al., 2011, 2012; Mészáros et al., 2018). The largest database of disorder annotation, MobiDB, provides a hierarchy of annotation based on the flavors and methods of disorder determination (in the order of experimental evidence from the DisProt database, indirect inference from secondary methods, and consensus prediction from computational methods; Potenza et al., 2015; Piovesan et al., 2016, 2017).
Due to the nature of these flavors, identifying the long-range overarching dependencies for disorder was a necessary step to consolidating accurate disorder prediction. An early method, Dispro, had incorporated sequential context through recursive NNs (Cheng et al., 2005; Hecker et al., 2008), but true whole-sequence learning was not applied until ESpritz (Walsh et al., 2012) trained three vanilla-BRNN-based methods on separate flavors of disorder. This method is still viable with its X-ray-based method (Espritz-Xray) ranked as the top-performing model by Necci et al. (2018), but does not provide a universal disorder flavor predictor.
The potential of an accurate and flavor-independent disorder predictor was realized by the simultaneous release of two contextual learning models: AUCpreD and SPOT-Disorder (Wang et al., 2016a; Hanson et al., 2017). Ranked as the top two disorder predictors in the review by Liu et al. (2017), AUCpreD utilizes an algorithm similar to the authors' previous work DeepCNF (described in a previous section) trained to maximize the area under the receiver operating characteristic curve (AUCROC), whereas SPOT-Disorder utilizes a deep LSTM-BRNN. Since then, a deep LSTM-BRNN has also been used for disorder prediction in NetSurfP-2.0 (Klausen et al., 2019). An update on SPOT-Disorder was proposed by utilizing an ensemble of LSTM-BRNNs and Inception-ResNets with Squeeze and Excitation connections (dubbed “IncReSenets”) to enhance propagation of dependencies both throughout the sequence and through the many layers of the network in SPOT-Disorder2 (Hanson et al., 2019). For comparison, SPOT-Disorder2 obtained a Matthews correlation coefficient (MCC; Matthews, 1975) of 0.499 in a subset of the dataset provided by Necci et al. (2018), compared to the 0.476, 0.434, and 0.462 obtained by Espritz-Xray, AUCpreD, and SPOT-Disorder, respectively (Hanson et al., 2019). The performances reported by these contextual algorithms are presented in Table 2, sorted by their publication date.
A Comparison of the Reported Area Under the Receiver Operating Characteristic Curve and Matthews Correlation Coefficient by Recently Released Contextual Protein Disorder Predictors and the Datasets the Results Were Obtained From
An asterisk (*) represents methods which do not utilize evolutionary profiles.
AUCROC, area under the receiver operating characteristic curve; MCC, Matthews correlation coefficient.
3.4. Single-sequence prediction of structural properties
Of note is that many of the predictors listed thus far depend on the use of evolutionary profiles derived from multiple sequence alignments (MSAs) generated by programs such as PSI-BLAST, HHBlits, HMMer, and MMseqs2 (Altschul et al., 1997; Finn et al., 2011; Remmert et al., 2012; Steinegger and Söding, 2017). These profiles provide information on the sequential homology of a protein to a target database of sequences. While these profiles boost the accuracy of these models for proteins with a sufficient number of effective sequences (Neff), the accuracy deteriorates for proteins with few homologs. Furthermore, evolutionary profile usage is prohibitive for large-scale analysis as searching extensive sequence datasets in MSA generation can take up to 99% of the total running time of a model.
Only several deep-neural-network (DNN)-based methods exist for single-sequence prediction of 1D structural properties, such as SPIDER-Single (Heffernan et al., 2018), PSIPRED (McGuffin et al., 2000), and single sequence-based DeepCNF (Wang et al., 2016b) for SS3 prediction. SPIDER-Single is the only predictor to utilize contextual learning through an iterative application of an LSTM-BRNN, requiring the network to learn the physicochemical residue intersequence dependencies during folding and not to rely on sequential homology between input sequences. This results in a drop in performance compared to its contemporary model SPIDER-3, except for low Neff proteins where the single sequence-based method excels. Predicting for a protein based on its sequential characteristics alone remains a fundamental question in the structural bioinformatics field.
Similar to secondary structure prediction, large genome-scale analysis of disorder is prohibited by the utilization of sequence profiles. Thus, there have been efforts to produce methods that perform well without the need for evolutionary profiles. Methods which analyze statistical potentials and amino acid propensities, such as IUPred (Dosztányi et al., 2005; Mészáros et al., 2018), GlobPlot (Linding et al., 2003b), and FoldIndex (Prilusky et al., 2005), naturally fall into this category. These are generally outperformed by single-sequence machine-learning based algorithms, such as the early PONDR series (Romero et al., 2001), DisEMBL (Linding et al., 2003a), MobiDB-Lite (Necci et al., 2017), single-sequence based AUCpreD (Wang et al., 2016a), CSpritz and single-sequence based ESpritz (Walsh et al., 2011, 2012), and SPOT-Disorder-Single (Hanson et al., 2018c). The two top-performing methods, Espritz and SPOT-Disorder-Single, both make use of recurrent connections to model intersequence dependencies. SPOT-Disorder-Single predicts for flavor-independent disorder, while Espritz performs single-sequence prediction for the same three flavors as its profile counterpart (X-ray, NMR, and DisProt annotations). Compared to the profile-based SPOT-Disorder, SPOT-Disorder-Single reported a >99% reduction in speed at a performance drop of 0.857 from 0.891 in AUCROC for a set of >11,000 proteins extracted from MobiDB.
4. PREDICTION OF PROTEIN CONTACT MAPS
A protein contact map is a 2D representation of a protein's folded structure, which details the residues of a protein which are in close proximity (i.e., in “contact”). This representation provides an informative constraint on the modeling of a protein's tertiary structure. The usefulness of predicted contact maps is illustrated by the fact that the alignment of contact maps is more effective at finding structural homologs than sequence alignment alone (Zhu et al., 2018). Early contact map predictors attempted to identify residues in contact by analyzing coevolving residues in protein MSAs, based on the underlying principle that residues in close proximity would jointly mutate according to the functional and structural needs of a protein (Göbel et al., 1994). While these evolutionary coupling analysis (ECA) methods were accurate for proteins rich with evolutionary information (Ovchinnikov et al., 2017), the performance deteriorated quickly for proteins with shallow MSAs due to a lack of homologs in a target database (Wang and Xu, 2013). Examples of this approach are CCMPred (Seemayer et al., 2014) and PSICOV (Jones et al., 2012).
Subsequent improvements were found in the application of SVM, deep belief networks (Hinton et al., 2006), and Recursive-NNs (Baldi and Pollastri, 2003), in predictors such as SVMSEQ (Cheng and Baldi, 2007), DeepConPred (Xiong et al., 2017), CMapPro (Di Lena et al., 2012), and NNcon (Tegge et al., 2009), respectively. Indeed, the winner of the 10th and 11th round of CASP found success using deep fully-connected NNs (Eickholt and Cheng, 2012; Jones et al., 2014). This improvement of these approaches over ECA was particularly noticeable for proteins with little evolutionary information, where higher-order correlations between residues learned by the network account for the limited homological information (Xu, 2018).
However, recent field analysis in the Critical Assessment of Structure Prediction (CASP) competitions (Schaarschmidt et al., 2018) has found that the propagation of contextual information has led to a far superior level of performance in the field than the previously listed approaches. The winner of CASP12, RaptorX-Contact (Wang et al., 2017), introduced the concept of treating protein contact map as an image segmentation task (Chen et al., 2018a), in which each protein residue pair is treated like a pixel in a 2D image. This is achieved through the use of an ensemble of deep 2D ResNets over a protein contact map where the channels are 1D (using outer concatenation) and 2D protein features. Similar approaches have since followed in DNCON2 (Adhikari et al., 2017), SPOT-Contact (Hanson et al., 2018a), DeepConPred (Ding et al., 2018), PConsC4 (Michel et al., 2018), DeepCov (Jones and Kandathil, 2018), DESTINI (Gao et al., 2019), and in Xu et al. (2018). SPOT-Contact improved on the architecture used in RaptorX-Contact by utilizing a method encapsulating a ResNet and a 2D variant of an LSTM-BRNN called a ReNet (Visin et al., 2015), in which a transposed LSTM-BRNN is applied over the y-dimension and then concatenated with another LSTM-BRNN operating over the x-dimension of the contact map. Meanwhile, DNCON2 predicted several interim contact maps at different separation thresholds ranging from 6Å to 10Å, which provide the input to a final model which predicts a final contact map at the typical 8Å threshold.
Another reason for the success of these methods is their usage of 2D features in the form of the outputs of ECA methods and contact potentials extracted from direct coupling analysis (DCA) on protein MSAs (Miyazawa and Jernigan, 1985; Betancourt and Thirumalai, 1999). Methods which combined the complementary outputs of several independent methods had already been established as high-performing predictors earlier in the literature (Jones et al., 2014; He et al., 2017), and so harnessing ECA methods for use in deep learning was seen as a lucrative method to exploit the strengths of both ECA and machine learning approaches. In doing this, the choice of sequence database for generating the evolutionary profiles and MSA used in each model gained importance in an attempt to provide as many potential homologs to input proteins as possible.
Ovchinnikov et al. (2017) proposed the use of metagenomics sequence libraries in protein modeling to greatly increase the size of available sequence databases. The use of these sequence libraries was shown to increase the number of proteins with a high Neff compared to UniRef100-2015 (Suzek et al., 2007) by a factor of 3.3, thus potentially increasing the number of sequences for which ECA methods are suitable for extracting evolutionary information. SPOT-Contact confirmed the importance of sequence database selection and ECA/DCA method inclusion by reporting a 1%–3% improvement in precision from their model by solely updating the sequence libraries by 1 year, as well as a 10% drop in performance on long-range contacts when trained without ECA/DCA information (Hanson et al., 2018a).
Recently, the contact map representation of proteins was extended to “distance” maps, where the binary classification of contact/not-in-contact was expanded to represent a distribution of selected distances (Xu, 2018; Zhu et al., 2018). This representation reduces the coarseness of the contact map, enabling a more refined conformational space with which to model a target protein's folded structure. This is exemplified by several high-performing entries in CASP13 (http://predictioncenter.org/casp13/doc/CASP13_Abstracts.pdf) adapting to this technique.
5. THE IMPACT OF CONTEXTUAL LEARNING ON TERTIARY STRUCTURE PREDICTION
The end goal of the prediction of these structural properties is to extract a reasonably accurate tertiary structure of a protein. The elements discussed in this article have gone through their own advances which in turn have formulated more effective tertiary structure models: secondary structure elements relate to the overall structural folding topology, protein backbone angles describe the local structure sequentially through the protein backbone, disorder denotes the lack of structure in proteins, and contact (or distance) maps frame the entire global structure into a constrained space. Thus, the ripples made in the literature by each incremental advancement of these subproblems are a confirmation of the divide and conquer nature of the protein structure prediction problem.
The CASP competition is a biannual analysis of not only the status of the protein structure prediction problem but also often the status of these related fields. Since its inception in 1994, CASP has provided blind testing to registered structure prediction groups for a snapshot of the current state-of-the-art performance. The entrants into CASP competitions for tertiary structure have generally been focused on the use of sequence-based alignments to template structures, typically using the protein primary and predicted, and ab initio modeling of protein using software suites such as Rosetta (Rohl et al., 2004), Modeller (Eswar et al., 2006), and I-TASSER (Zhang, 2008). Template matching approaches can be quite accurate only if the target protein is matched with a template from the same fold (as defined in SCOP or CATH; Murzin et al., 1995; Orengo et al., 1997; Yang et al., 2011). However, the scoring of the sequential alignment can be unreliable especially for proteins without a known fold or suitable reference in the template library. In contrast, ab initio methods have been shown to produce native conformations for input sequences through simulated annealing optimization when provided with an optimal energy function (Yang and Zhou, 2016). Since there is no such globally-optimal function available this approach was typically limited to the modeling of small protein fragments.
These techniques saw a rather stagnant pattern appear for over 10 years as computational models only managed small incremental improvements in each iteration of development. This trend between CASPs was broken in the 11th and 12th rounds, which saw the introduction of accurate contact map prediction models through powerful machine learning coupled with coevolution information, greatly enhancing the performance of both template matching and ab initio models using predicted contacts to constrain their tertiary model (Moult et al., 2016, 2018). This was especially the case for challenging free-modeling (FM) targets for which no reliable template structure is available, presenting the first time that long, novel protein chains could be somewhat accurately modeled. This breakthrough stemmed from novel applications of ab initio methods, particularly by the incorporation of empirical potentials based on predicted structural properties, such as protein contact maps, to guide their folding simulations rather than the classical approach governed by physics-inspired empirical and/or knowledge-based energy functions (Hou et al., 2019).
The most recent edition of CASP (CASP13 in 2018) epitomizes the expressiveness and potential of contextual learning for the modeling of important structural characteristics and its effects on protein structure prediction. Following the success of RaptorX-Contact in CASP12 (Wang et al., 2017), global context modeling has become imperative for ensuring a competitive entry in the standings. Entrants in CASP13 have introduced contextual modeling in the form of ResNets, residual LSTM networks (Kim et al., 2017), fully-CNNs (Long et al., 2015), and 2D Recursive-NNs (Baldi and Pollastri, 2003). This competition also witnessed an unprecedented boost in the accuracy for FM targets due to the use of metagenomics libraries for coevolution analysis (Ovchinnikov et al., 2017), greatly enhancing the ECA component of contact map predictions.
In the tertiary structure and contact map prediction categories, AlphaFold (A7D) and RaptorX-Contact (hereafter dubbed “Raptor-2018” to distinguish between the previous iteration), respectively, both managed to effectively leverage the use of sequential information with contextual learning to maximize the power of their models. The overall best method for tertiary structure prediction, AlphaFold (Evans et al., 2018), generated tertiary structure through the combination of deep-learning predicted contact and angle restraints with fragment sampling by the Rosetta suite (Rohl et al., 2004). Rosetta was used as an iterative tuning environment to optimize the predicted fragments according to a scoring function based on the predicted inter-residue distance map, itself generated by a deep ResNet. The initial fragments were constructed by the predicted backbone torsion angles from an RNN variant. This approach can be seen as an amalgamation of old physics-based approaches and the current wave of contextual deep learning, as it minimizes a protein-specific knowledge-based potential based on the predictions of a set of incredibly effective NNs.
A similar pipeline has also been proposed by Ingraham et al. (2019) for end-to-end differentiation for NN-based protein-specific energy function prediction. Unfortunately, the performance of this model was not benchmarked in CASP13. AlphaFold, along with the next-best prediction groups, which utilized contact maps as input to the I-Tasser suite (Zhang, 2008) and CONFOLD2 (Hou et al., 2019; MULTICOM), illustrates the benefit of contextual-based machine learning to extract more meaningful protein representations.
For protein contact map prediction, Raptor-2018 utilizes a similar approach to its previous iteration, but utilizes an ensemble of deep ResNets to predict a distance map rather than a contact map. Interestingly, the summation of the predicted probabilities of the residue pairing being <8Å gives rise to a 2% increase in accuracy over the standard contact map prediction (Xu, 2018). As an illustration, the Raptor group's predicted contact map and tertiary structure for protein T0957s2-D1 from CASP13 are shown in Figure 4.

The contact map
One final aspect of protein tertiary structure prediction is the method for selecting a near-native structure from a host of generated model structures. Model quality assessment has typically relied on the use of handcrafted features based on the atomic coordinates as input to fully-connected NNs, SVMs, or other probabilistic methods (Bhattacharya et al., 2016; Manavalan and Lee, 2017; Uziela et al., 2017). Recent applications of 3D-CNNs have shown that contextual modeling can successfully be applied to identify accurate tertiary structure models (Derevyanko et al., 2018; Pagès et al., 2019) using minimal sequence features. However, moderate performance of these methods in CASP13 Quality Assessment shows that further refinement is required to match the performance of noncontextual methods.
6. NOTES AND THOUGHTS FOR THE FUTURE
6.1. Machine learning architectures
The forefront of machine learning is constantly changing. New architectures, whether completely novel or slight variants on existing methods, are rapidly being proposed along with new benchmarks to be beaten. Several of these methods have not yet seen widespread application in protein structure prediction despite their applicability to protein data. One major application is the use of attention-based networks, which can identify relevant sequential components through the use of context vectors calculated over the entire sequence for each data point (Vinyals et al., 2015). This is particularly promising for protein data due to its potential for identifying nonlocal interactions for specific residues within the sequence. Recent iterations of attention mechanisms, such as the Transformer network (Vaswani et al., 2017) and Pervasive Attention (Elbayad et al., 2018), have offered vastly different approaches to identify significant sequence components and have displayed a great aptitude in sequence processing for machine translation tasks.
Another promising technique is the use of dilated convolutions as a simple way to improve the contextual field of a CNN without increasing the parameter count. This method has reported to improve on the performance of RNN-based techniques (Bai et al., 2018), which is significant as CNNs have a greatly reduced parameter count and computational load compared to RNN architectures. Dilated convolutions have already been used for protein contact map prediction in several CASP13 participants (such as AlphaFold and Raptor-2018), as well as in Xu et al. (2018), which gave an immediate 2%–4% boost to long-range precisions over standard CNN kernels. Finally, Google's recent NasNet article (Zoph et al., 2018) shows the potential for automated model selection, removing the onus of hyperparameter and model selection for contextual learning from researchers. In this architecture, a NN called a “controller” proposes a separate NN architecture for training on an objective. The controller then receives the accuracy of the proposed architecture as feedback to improve its next proposal using reinforcement learning. While training these models on workstations available to standard researchers may perhaps currently be unfeasible, steps are being made to decrease the necessary computational power for quick training (Liu et al., 2018).
6.2. Protein structure prediction
One drawback of the state-of-the-art machine learning models is that novel or underrepresented folds in the sequence dataset are often poorly predicted due to their low-quality evolutionary profiles derived from few (if any) homologous sequences. As stated previously, the use of evolutionary profiles has led to a large increase in many protein structure prediction problems, but for the vast majority (>90%) of proteins with few homologous sequences (Ovchinnikov et al., 2017) this is not the case. The core of Anfinsen's dogma, that the folded globular form of a protein is solely encoded in its primary sequence, theorizes that protein structure prediction should not strictly require the use of evolutionary information. Extracting this information without the use of sequence profiles, however, is still lacking, illustrated by the drop in performance for secondary structure (11% in SS3) and protein disorder prediction (0.06 in MCC) between the latest single-sequence predictors and their profile-based counterparts. However, these methods are still sought after despite the accuracy discrepancy due to their viability for large-scale analysis and their contribution in achieving the ultimate goal of solving the relationship between a single sequence and its structure.
One possibility to realizing true end-to-end protein structure prediction is to train and predict 3D structure directly by machine learning without the use of molecular modeling frameworks. These frameworks, despite their usefulness in generating accurate structures, may inadvertently introduce some human bias due to the use of handcrafted scoring techniques, often regarding the assumptions made in the physics-inspired energy functions. Indeed, many landmark machine learning applications, such as AlphaGo and AlphaZero, are allowed to train without human influence to gain latent understanding of the problem at hand. In protein structure prediction, if we obtain a large enough dataset, there is a possibility that the unknown energy function behind protein folding can be learned to fold a sequence into the right conformation without the need for energy optimization. Recently, Al Quraishi (2019) showed such a possibility using predicted backbone angles to construct 3D structures and minimizing the root-mean-square distance deviation between the constructed model and the native structure, all within an LSTM-BRNN model framework, dubbed recurrent geometric networks. This new approach, however, can produce unphysical conformations with forbidden backbone angles and overlapped atoms. How to incorporate these physical restraints will be key in solving the protein structure prediction problem by machine learning alone.
Another work by Ingraham et al. (2019) applies a somewhat similar pipeline to AlphaFold, but applies a Langevin dynamics simulation to enable end-to-end differentiation of the protein structure, marking a continuous sequence-to-structure pipeline with joint optimization of the free-energy minimizer and NN layers. This is unlike AlphaFold, which has separate training of its scoring and energy optimization.
The core of protein structure prediction has been concerned with the localization of the central Carbon-α atom for each residue in the primary sequence. However, amino acid side chains, which dictate the physicochemical reactions in the folding path, are often not incorporated into these models despite their structural and functional significance. Residue side chain position has important functional impact in protein–protein docking in protein complexes (Gray et al., 2003; Wang et al., 2005) and also has implications for sampling protein tertiary structure conformations (Krivov et al., 2009). This is a separate challenge of protein structure refinement (Feig, 2017).
Finally, the end objective of this research is to understand the function of proteins. The focus on deriving structure stems from the structure-function dogma (with disorder considered as an extension to this tenet), which states that protein structure directly determines the function of a protein (Dunker and Obradovic, 2001). Following the advances discussed in this work, it is natural to assume that the same methods would apply to the problem of residue-level and protein-level function prediction. Historically, a major limitation of this strategy has been the availability of annotated functional/nonfunctional protein data required for training deep contextual models. Several approaches utilizing noncontextual approaches for predicting protein functionality have been implemented to fill this gap (Taherzadeh et al., 2016; Fa et al., 2018); however, these approaches have limited expressiveness relative to deep contextual models. More recently, shallow contextual models have been developed for drug-target binding affinity (Öztürk et al., 2018; Lee et al., 2018). These models are only limited in their contextual modeling due to only utilizing several convolutional layers. These fields are likely to undergo contextual modeling as more data become available.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Australian Research Council DP180102060 to Y.Z. and K. P. and, in part, by National Health and Medical Research Council of Australia 1121629 to Y.Z. The authors also gratefully acknowledge the use of the High Performance Computing Cluster “Gowonda” to complete this research and the aid of the research cloud resources provided by the Queensland Cyber Infrastructure Foundation.
AUTHOR DISCLOSURE STATEMENT
The authors declare there are no competing financial interests.