
Abstract
Dipeptidyl peptidase-4 (DPP-4) is considered a major drug target for type 2 diabetes mellitus (T2DM). In addition to T2DM, a regulatory role of DPP-4 was also found in cardiovascular diseases. Existing DPP-4 inhibitors have been reported to have several adverse effects. In this study, a computer-aided drug design approach and its use to detect a novel class of inhibitor for DPP-4 are reported. Through structure and pharmacophore-based screening, we identified 13 hit compounds from an ∼4-million-compound library. Physical interactions of these hits with DPP-4 were studied using docking and explicit solvent molecular dynamics (MD) simulations. Later, MMPBSA binding energy was calculated for the ligand/protein simulation trajectories to determine the stability of compounds in the binding cavity. These compounds have a novel scaffold and exhibited a stable binding mode. “Best-in-screen” compounds (or their closest available analogs) were resourced and their inhibition of DPP-4 activity was experimentally validated using an in vitro enzyme activity assay in the presence of 100 and 10 μM compounds. These assays identified a compound with a spirochromanone center with 53% inhibition activity at a 100 μM concentration. A further five spirochromanone compounds were synthesized and examined in silico and in vitro; again, one compound showed 53% inhibitory activity action at 100 μM. Overall, this study identified two novel “spirochromanone” compounds that lowered DPP-4 activity by more than ∼50% at 100 μM. This study also showed the impact of fast in silico drug design techniques utilizing virtual screening and MD to identify novel scaffolds to bind and inhibit DPP-4. Spirochromanone motif identified here may be used to design molecules to achieve drug-like inhibitory action against DPP-4.
Introduction
Diabetes mellitus (DM) is likely one of the oldest known diseases; it was first reported in an Egyptian manuscript nearly 3000 years ago (Tattersall, 2010). In the early twentieth century, two forms of this chronic disorder were distinguished and designated as type 1 and type 2 DM (Himsworth, 1936), with the latter also known as metabolic syndrome (Patlak, 2002), hyperglycemia, insulin resistance, and relative insulin deficiency. Whereas type 1 DM is caused by a deficiency in insulin production, type 2 DM (T2DM) sufferers do not respond to insulin and, in later stages of the disease, may also suffer from decreased insulin production. T2DM is the most common form of diabetes and many studies have confirmed the contribution of a variety of genetic, environmental, and behavioral factors to its onset (Chen et al., 2012). According to the World Health Organization, the global disease burden of diabetes in adults has risen from 4.7% in 1980 to 8.5% in 2014 (Mathers and Loncar, 2006).
Insulin is a small peptide hormone released by pancreatic beta cells in response to elevated glucose levels in the blood. Primarily, insulin triggers cellular glucose uptake, but it is also involved in regulating carbohydrate and lipid metabolism by promoting glucose storage in the form of glycogen, lipids, and protein. Furthermore, insulin also stimulates the cellular uptake of amino acids, inhibits protein degradation, and promotes protein synthesis (Joost, 2011). The ubiquitously expressed cell surface protease dipeptidyl peptidase-4 (DPP-4) suppresses the insulinotropic effects of the insulin regulator incretin hormone glucagon-like peptide (GLP-1) through proteolytic degradation of GLP-1 (Gerich, 1998). In line with this molecular mechanism, DPP-4-deficient mice showed improved glucose tolerance and higher plasma concentrations of the incretin glucose-dependent insulinotropic peptide, GLP-1, and insulin (Proud, 2006). Owing to its hyperglycemic activity, GLP-1 has been used extensively for the treatment of T2DM (Barnett, 2006) and, following validation of DPP-4 as a target for control of T2DM, DPP-4 inhibitors have been in clinical use as antidiabetic therapeutics since their action prolongs the activity of endogenously released GLP-1 (Proud, 2006; Shirakawa et al., 2016).
In addition to the therapeutic value of DPP-4 for T2DM, inhibition of this enzyme has also been shown to elicit beneficial effects in cardiovascular diseases, including atherosclerosis and ischemic heart disease, the leading cause of premature adult mortality worldwide (Lusis, 2000). Whereas the detailed biological mechanisms behind atherosclerosis remain unknown, various studies suggest that some DPP-4 inhibitors also possess cardioprotective effects (Chinda et al., 2013; Hausenloy et al., 2013; Ferrannini and DeFronzo, 2015; Gilbert and Krum, 2015; Zhong et al., 2015; Kubota et al., 2016), including beneficial outcomes for atherosclerosis sufferers (Remm et al., 2015; Silva Júnior et al., 2015; Singh et al., 2015). Accordingly, DPP-4 is at present also considered a potential drug target for chronic arterial diseases.
The intrinsic membrane glycoprotein DPP-4 is a serine protease with 766 amino acids and its structural topology comprises (1) a cytoplasmic tail, (2) a transmembrane domain, and (3) an extracellular domain. The extracellular domain is the most relevant for therapeutic intervention because it is composed of the catalytic site and an eight-bladed β-propeller domain that is connected to the catalytic domain (Thoma et al., 2003). Mechanistically, DPP-4 is an exopeptidase that cleaves proline-containing dipeptides from the N-terminus (Röhrborn et al., 2015); proteolysis occurs in the catalytic domain, using a catalytic triad of Ser630, Asp708, and His740. Among the first inhibitors of DPP-4 were saxagliptin (Augeri et al., 2005) and vildagliptin (Mathieu and Degrande, 2008), which target the substrate binding subsites (S1 and S2) of DPP-4. Accordingly, residues of interest for drug discovery include Val656, Trp662, Tyr666, and Val711 (S1 pocket); Arg25, Phe357, Arg358, Tyr547, Pro550, and Asn710 (S2 pocket); and residues in the N-terminal recognition region formed by Glu205, Glu206, and Tyr662 (Arulmozhi and Portha, 2006). Subsequently, optimized inhibitors with improved selectivity and lesser side effects have been developed, including alogliptin (Feng et al., 2007), anagliptin (Kato et al., 2011), evogliptin (Kim et al., 2012), gemigliptin, linagliptin (Eckhardt et al., 2015), omarigliptin (Biftu et al., 2014), sitagliptin (Thornberry and Weber, 2007), and teneligliptin (Yoshida et al., 2012a).
Whereas DPP-4 inhibitors in general are tolerated well by patients, they can provoke hyperglycemia (Salvo et al., 2016), heart failure (Dicker, 2011), and acute pancreatitis (DeVries and Rosenstock, 2017) when used in combination with sulfonylurea derivatives and insulin (Salvo et al., 2016). There are also reports of nasopharyngitis, headache, upper respiratory tract infection, and urinary tract infection (Ligueros-Saylan et al., 2010; Ali and Fonseca, 2013), and thus, while existing inhibitors are potent against DPP-4 (IC50 values in the nanomolar inhibition range) (Berger et al., 2018), they have potentially severe side effects in what is already a vulnerable patient group. Therefore, work is ongoing to identify a new chemical scaffold for inhibiting DPP-4 with less or no adverse effects for patients.
The availability of experimental three-dimensional (3D) structures of the ligand-bound DPP-4 extracellular domains (Feng et al., 2007; Edmondson et al., 2009; Yoshida et al., 2012a, b; Nabeno et al., 2013) allows the application of computational methodologies to discover and design new inhibitors for this therapeutic target, particularly work that aims to discover new hit compounds that can be translated into chemotherapeutics with minimal side effects. In this study, we report an in silico drug design approach to identify new hit compounds to bind and potentially inhibit DPP-4. Following screening of a large compound library (N ≈ 4 million), 50 hits were selected based on the favorability of their predicted interactions and docking energies. The selected molecules were docked and subjected to molecular dynamics (MD) simulations as potential DPP-4:ligand complexes to calculate the binding energies with Poisson-Boltzmann surface area continuum solvation (MM/PBSA). The dynamics behavior of each compound in the substrate binding cavity of DPP-4 was also examined. A total of 13 compounds were selected and sourced commercially and a small library of five readily available synthetic compounds with similar topology was generated. The resulting set of 17 compounds was subjected to an in vitro enzyme inhibition assay.
Methods
Earlier studies divided the binding pocket of DPP-4 into two spatial regions labeled the S1 and S2 (Arulmozhi and Portha, 2006) sites. However, an additional binding region was also identified that covers the N-terminal recognition site. Cocrystal structures of known inhibitors with DPP-4 were used here as reference molecules to locate these binding regions: alogliptin, anagliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin (PDB 3GOB, 3WQH, 2RGU, 3BJM, 4FFW, and 3W2T, respectively) (Eckhardt et al., 2007; Metzler et al., 2008; Robert et al., 2009; Tang et al., 2012; Nabeno et al., 2013; Watanabe et al., 2015). Overall, this study utilized three rounds of virtual screening to identify potential hit compounds. Runs 1 and 2 utilized a protein structure-based approach, while Run 3 was a ligand-based approach using pharmacophores of known inhibitors to search the ZINC database. Compounds identified from all three rounds were further examined by docking into the DPP-4 active site, 20 ns MD simulations and MMPBSA binding energy calculations. These highest scoring compounds were then tested for inhibition of DPP-4 in vitro. Compounds were resourced from MolPort (Latvia) where they are further supplied by ChemDiv, Inc., ENAMINE Ltd., Pharmeks, Ltd., Specs and TargetMol, and Vitas-M Laboratory, Ltd. via MolPort.
Screening and identification of hit compounds
Virtual screening Runs 1 and 2 used the HitGen module of commercial drug design tool INVENTUS™ (www.novoinformatics.com/inventus.php) for structure-based screening, which uses physicochemical features to screen compounds from an inbuilt compound library consisting of ∼4 million compounds (Mukherjee and Jayaram, 2013). The physicochemical filters for screening were estimated from the computed values of known inhibitors (alogliptin, anagliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin) selected previously as reference compounds. These physicochemical filters were as follows: (1) a number of hydrogen bond donors (HBD), (2) a number of hydrogen bond acceptors (HBA), (3) logP, (4) a number of aromatic atoms, (5) a number of atoms, (6) carboxyl group count, (7) hydroxyl group count, (8) phosphate/sulfate group count, (9) amide group count, (10) ketone/aldehyde group count, and (11) ester/ether group count. Run 1 did not use any organic functional group filter criteria to screen compounds, while Run 2 explicitly used a count for these functional groups, estimated from their corresponding values in known inhibitors.
Table 1 shows the list of physicochemical filters used in screening and their respective ranges for Runs 1 and 2. The top 50 hit compounds from each run were identified using the scoring function of the screening protocol implemented in INVENTUS (Mukherjee and Jayaram, 2013). These hit compounds were docked into the binding site of DPP-4 (identified using the vildagliptin-DPP-4 structure; PDB 3W2T) using the INVENTUS docking module (NovoDocker), which applies a rigid docking grid search technique (Gupta et al., 2007). The binding energy of each docked pose generated was calculated using an all-atom energy-based computational protocol incorporated in the BioAff module of INVENTUS (Jain and Jayaram, 2005). The docking program generates the best eight poses for each protein/ligand complex and those compounds that had negative binding energy across all docked poses were selected for further analysis. Screening, docking, and binding energy analysis resulted in the selection of six compounds from Run 1 and five compounds from Run 2 from the top 50 compounds in each round.
Parameters Used for Screening in Run 1 and Run 2 Under INVENTUS Package with Their Respective Filtering Range to Generate Top 50 Hit Compounds
Parameters Used for Screening in Run 1 and Run 2 Under INVENTUS Package with Their Respective Filtering Range to Generate Top 50 Hit Compounds
HBA, hydrogen bond acceptors; HBD, hydrogen bond donors; NF, no filters applied on given parameter.
In screening Run 3, the PharmaGist (Schneidman-Duhovny et al., 2008) server was used to generate a three-point pharmacophore from known inhibitors of DPP-4 (alogliptin, anagliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin). These known drugs share similar geometrical arrangements of one HBD, one HBA, and one aromatic ring, and the resulting pharmacophore was applied using the ZincPharmer (Koes and Camacho, 2012) server to screen the ZINC compound database. This produced 2000 compounds, which were further ranked and screened against the DPP-4 protein using the custom library option in the HitGen module of INVENTUS. In this step, the pharmacophore 2000 was used as a search library for structure-based screening in INVENTUS as in Runs 1 and 2. Again, the top 50 hits were docked using NovoDocker and compounds with negative binding energy across all docked poses selected for further analysis. This process selected two compounds from Run 3. In total, the 13 compounds selected from the three screening runs were redocked using AutoDock version 1.5.6 (Morris et al., 2009) to cross-validate their predicted binding affinities for DPP-4. The highest ranked docked pose for each molecule was analyzed by the stand-alone LIGPLOT tool to sketch an interaction plot (Wallace et al., 1995). Last, the interacting residues shown in LIGPLOT for selected protein/ligand complexes were compared with the residues of existing binding sites: S1, S2, and the N-terminal recognition region. A detailed, stepwise explanation of the INVENTUS and the AutoDock parameters used is provided in Supplementary Material.
GROMACS 4.6.4 (Berendsen et al., 1995) was used to simulate the highest scoring docked pose of DPP-4 protein with each compound (both screening hits and purchased analogs thereof). These complexes were simulated for 20 ns in an 11.9 × 11.9 × 11.9 Å3 water box under explicit solvent conditions. Initial complexes were energetically minimized with solvent using 5000 steps of a steepest descent algorithm. Postminimization, the complete system was equilibrated for 100 ps and 1 ns under NVT (canonical ensemble) and NPT (isothermal isobaric ensemble) ensembles, respectively. The NVT equilibration ensemble used velocity rescale as temperature coupling, while NPT equilibration had velocity rescale as temperature coupling and Berendsen pressure coupling. During each 20 ns simulation phase, pressure coupling was changed to Parrinello-Rahman. Root mean square deviation (RMSD) and hydrogen bond counts were computed for each 20 ns trajectory using g_rms and g_hbond. The MMPBSA binding energies of DPP-4 with each compound were computed using g_mmpbsa (Kumari et al., 2014), which calculates the nonpolar solvation energy of each protein/ligand complex using the solvent accessible surface area model. This tool allows prediction of molecular interactions comprising both van der Waals forces and electrostatic interactions with polar and nonpolar solvation terms for each simulation frame. During the calculation of polar solvation energy, the solvent dielectric constant was fixed at 80; the protein dielectric constant was set at 1. The linear Poisson/Boltzmann equation was used to solve the continuum solvent model (Fogolari et al., 2002).
DPP-4 inhibition assays
DPP-4 inhibition was assessed using a DPP-IV Drug Discovery Kit (BML-AK499; Enzo Life Sciences, Farmingdale, NY). All assays were performed at 37°C, in triplicate, according to the kit protocol. Compounds were made up in dimethyl sulfoxide (DMSO) and added such that the final concentration of DMSO was 1%. Controls included DPP-4 enzyme with either buffer (uninhibited) or 10 μM P32/98 (control inhibitor) added. To test compounds for DPP-4 inhibition, 2.5 nM DPP-4 was first incubated with 10 μM AMI compounds 1–11 for 10 minutes. The reactions were then initiated by the addition of 5 μM H-Gly-Pro-pNA substrate and continuously monitored through the absorbance of the pNA product at 405 nm for 35 minutes. The data were processed for a time window of 0–31 minutes: initial rates were obtained from linear fits in R, scaled relative to an uninhibited enzyme control, and plotted with error bars showing the standard error. Significance was assessed using the Student's t-test. Those compounds that showed some inhibition, reducing the relative DPP-4 activity by at least 15%–20%, were retested at 100 μM, as per the lowest concentration used by Guasch et al.'s (2012) work.
Results and Discussion
Virtual high-throughput screening (vHTS) is an established technique for rapid screening of large sets of compounds against a given protein target to identify potential binding molecules (Subramaniam et al., 2008). The core principle of this methodology is based on the physicochemical complementarity between a ligand and the defined binding pocket of a protein (Subramaniam et al., 2008). In this study, vHTS was used to screen the ZINC (Irwin et al., 2012) and NCI (Ihlenfeldt et al., 2002) compound libraries (4 million molecules) to discover novel potential inhibitors. This work used the crystal structure of DPP-4 complexed with vildagliptin (PDB 3W2T) (Mathieu and Degrande, 2008) as a reference. Based on previously characterized interactions between DPP-4 inhibitors and various residues in the substrate binding site, a set of filters was generated to guide selection of hit compounds. The filters were designed to consider ligand interactions with the S1 and S2 subsites, as well as the N-terminal recognition region of DPP-4, and were implemented as a quantitative structure/activity relationship equation in the molecular modeling procedure (Mukherjee and Jayaram, 2013). Two independent vHTS campaigns were conducted, each applying a different set of filter criteria (see details in the Methods section). In addition, an independent vHTS was performed using a pharmacophore search method. From the three vHTS campaigns, 50 compounds were selected based on virtual screening scores and estimated protein/ligand binding energies (Mukherjee and Jayaram, 2013).
Recently, several empirical binding energy equations have been proposed to calculate improved estimates of the ligand affinity of a particular protein (Kollman et al., 2000; Schulz-Gasch and Stahl, 2004). Thus, we used the NovoDocker (Gupta et al., 2007) and BioAff (Jain and Jayaram, 2005) modules of the INVENTUS package to further appraise interactions between the 50 selected compounds and DPP-4. Eight binding poses were generated for each of the 50 compounds and binding energy scores were calculated using the INVENTUS scoring function (Jain and Jayaram, 2005). For compounds that yielded negative binding energies for all eight poses (n = 13), protein/ligand complexes were generated using the complex of vildagliptin with DPP-4 as a guide (Mathieu and Degrande, 2008) and subjected to MD simulations extending over 20 ns, with subsequent calculation of free binding energies using MM/PBSA (Kollman et al., 2000). Of the selected 13 compounds, 12 could be sourced commercially (although some of the final compounds purchased were analogs of the original hits with a minimum similarity of 70% [Tanimoto coefficient]).
The analogs were tested in silico against the same docking and MD simulations as their parent compounds. The purchased compounds were tested for inhibition of DPP-4 in an in vitro enzyme assay at concentrations of 10 and 100 μM. A small library of five readily available compounds exploring the recurring spirochromanone-type scaffold was synthesized in-house and included in the enzyme inhibition assays. These five compounds were also subjected to molecular docking and simulations to predict their behavior in the DPP-4 binding pocket.
Virtual screening using DPP-4 inhibitor-derived filters
In two independent screening runs (Run 1 and Run 2), different sets of filters were applied in vHTS of the large compound library using the screening module of INVENTUS. From each run, the top 50 compounds, as ranked by vHTS protein/ligand complementarity scores (Mukherjee and Jayaram, 2013), were docked into the binding site, which was predicted using the cocrystal structure of the reference ligand vildagliptin and DPP-4 (PDB 3W2T). Eight binding poses were generated for each protein/ligand pair (Supplementary Table S1). Compounds that yielded a negative docking score for all eight poses were selected to be carried forward. This criterion resulted in the selection of six (
These compounds have logP values in the range of 2.14–4.06 and molecular masses of 387–483 Da. Notably, all 11 hit compounds possess two or more aromatic rings, which is a recurring pharmacophore in many known DPP-4 inhibitors (alogliptin, anagliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin) (Agrawal et al., 2012). To assess the structural diversity of the identified hits, the Tanimoto coefficient (Bajusz et al., 2015) was used as a standard measure of similarity between pairs of compounds. A score below 0.5 is considered an acceptable cutoff for low similarity (Baldi and Nasr, 2010). Among all 56 pairings, a maximum score of 0.6 was obtained for compounds
Pharmacophore screening
In addition to screening using the custom-generated filters, pharmacophore-based screening was also performed (Run 3). Conceptually, 3D pharmacophores are generated based on the structures of known ligands (Moro et al., 2005; Gao et al., 2010; Amaravadhi et al., 2014). In this study, a three-point pharmacophore was generated using the structures of six known DPP-4 inhibitors (alogliptin, anagliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin) and used to search the ZINC database, which resulted in the identification of 2000 compounds (Supplementary Table S3). Subsequently, this customized library of 2000 compounds was used in vHTS with the INVENTUS screening module (Mukherjee and Jayaram, 2013). As in Runs 1 and 2, the top 50 compounds were selected based on protein/ligand complementarity scores and subjected to docking calculations with the same DPP-4 binding site as described above. Eight poses for each protein/ligand pair were generated and assessed for their binding energies. Two molecules (
Virtual screening summary
Using the virtual screening protocol described above, a set of 13 diverse compounds with high predicted binding scores was identified. The diversity of the compounds within the set and when compared with six established DPP-4 inhibitors is illustrated in the hierarchical clustering similarity matrix in Figure 1a, where 0 represents no similarity and 1 indicates 100% similarity.
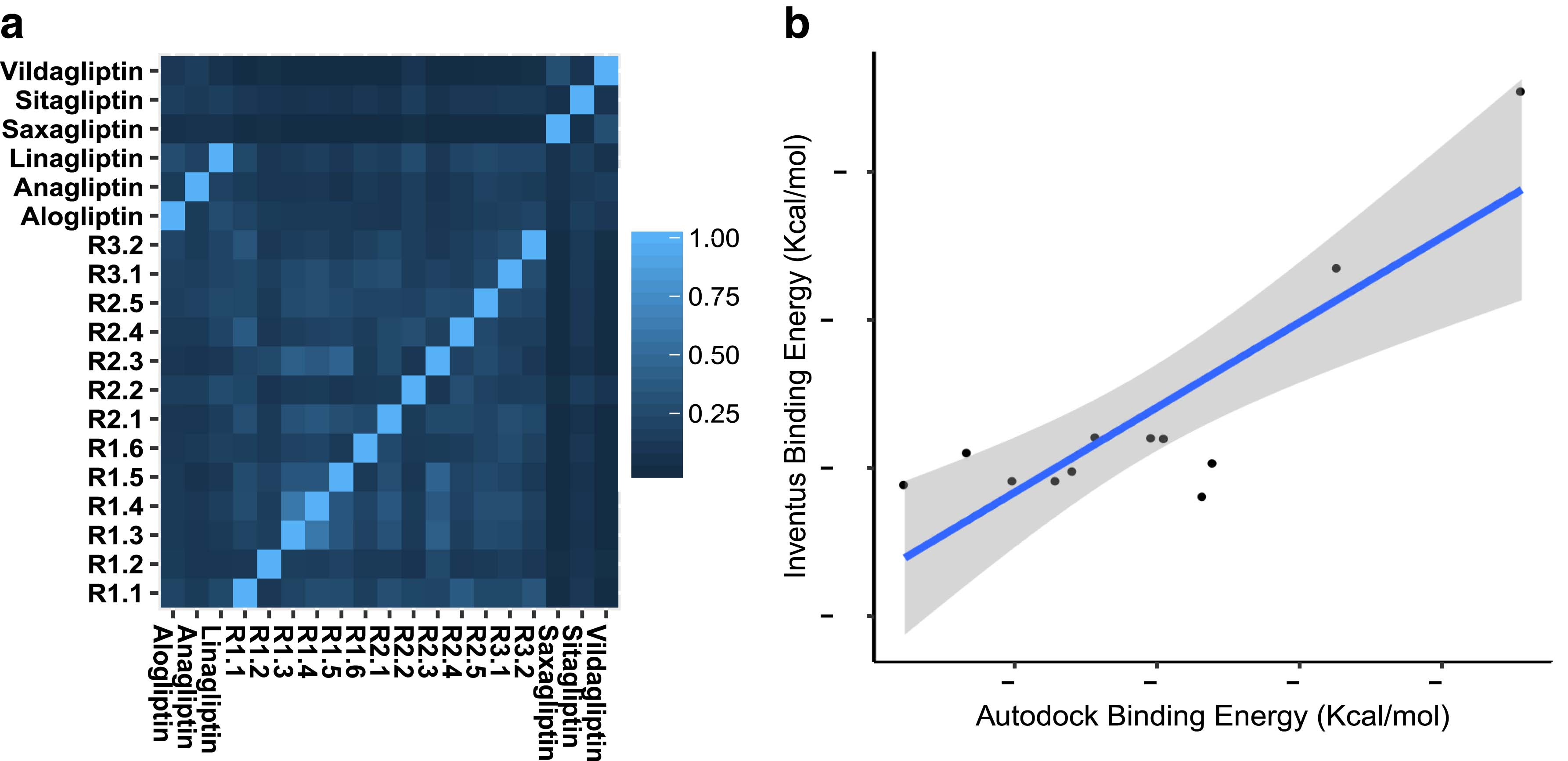
Similarity index and consistency of predicted binding scores for compounds identified by virtual screening.
Correctly distinguishing between binding and nonbinding compounds is a known challenge in virtual screening studies (Deng et al., 2014), owing to the potential shortcomings of the scoring function implemented within a particular software package. Therefore, we sought to corroborate the binding scores derived from INVENTUS by docking the 13 identified compounds using AutoDock (Morris et al., 2009). The calculated binding energies obtained with INVENTUS ranged between −12.37 and −6.91 kcal mol−1; for AutoDock, the corresponding binding energy range was from −11.77 to −3.45 kcal mol−1. Notably, the compounds identified by pharmacophore search (Run 3) exhibited lower binding scores than compounds identified with DPP-4 inhibitor-derived filters (Runs 1 and 2). The best binding scores for each compound from each program were analyzed for correlation (Fig. 1b) and yielded a Pearson correlation coefficient of 0.84, indicating consistency in the predicted binding scores. Protein/ligand interactions in the highest scoring pose for each compound docked into the DPP-4 binding site using AutoDock are illustrated in Supplementary Figure S1.
As mentioned, the vildagliptin-DPP-4 cocrystal structure (PDB 3W2T) was used to select and define the binding region for this study. In AutoDock, a cube (grid points 52, 72, 50) was created around the reference ligand (vildagliptin) to dock vHTS compounds. The interacting residues from within this defined binding region of DPP-4 in the highest scoring docked pose of each compound are largely similar, as shown in Supplementary Figure S1. These interactions are dominated by hydrophobic contacts (shown in dotted red lines) supported by hydrogen bonds (green dotted lines), which likely strengthen binding.
Molecular dynamics
To overcome the lack of solvation in docking procedures, MD simulations with explicit waters are frequently applied as a subsequent assessment in the course of computational drug discovery (Alonso et al., 2006; Sixto-López et al., 2017). With present computational resources, MD is typically performed with a simulation time of 20 ns in this context (Gurung et al., 2016; Dolatkhah et al., 2017). In addition to the evaluation of conformational changes in the protein and ligand in the presence of solvent and the potential contribution of solvent-mediated interactions, MD simulations also allow the calculation of improved binding energies by using Poisson/Boltzmann surface area continuum solvation (MM/PBSA) (Brown and Muchmore, 2007; Valiev et al., 2008). Using the highest scored docked poses of the 13 compounds (identified based on the vildagliptin binding site; PDB 3W2T) as the starting conformations, MD simulations and MM/PBSA binding energy calculations were performed for 20 ns. A pre-equilibration MD was run for 100 ps and 1 ns under an NPT ensemble to compensate the effect of the added solvent. For comparison, the structure of the DPP-4:vildagliptin complex (PDB 3W2T) (Mathieu and Degrande, 2008) was subjected to the same process.
Conformational stability
The conformational flexibility of each ligand was evaluated by monitoring the RMSD (Cau et al., 2015) of its structure for the duration of the MD simulation (see Fig. S2a–c). The Cα-RMSD data indicate that all compounds with the exception of
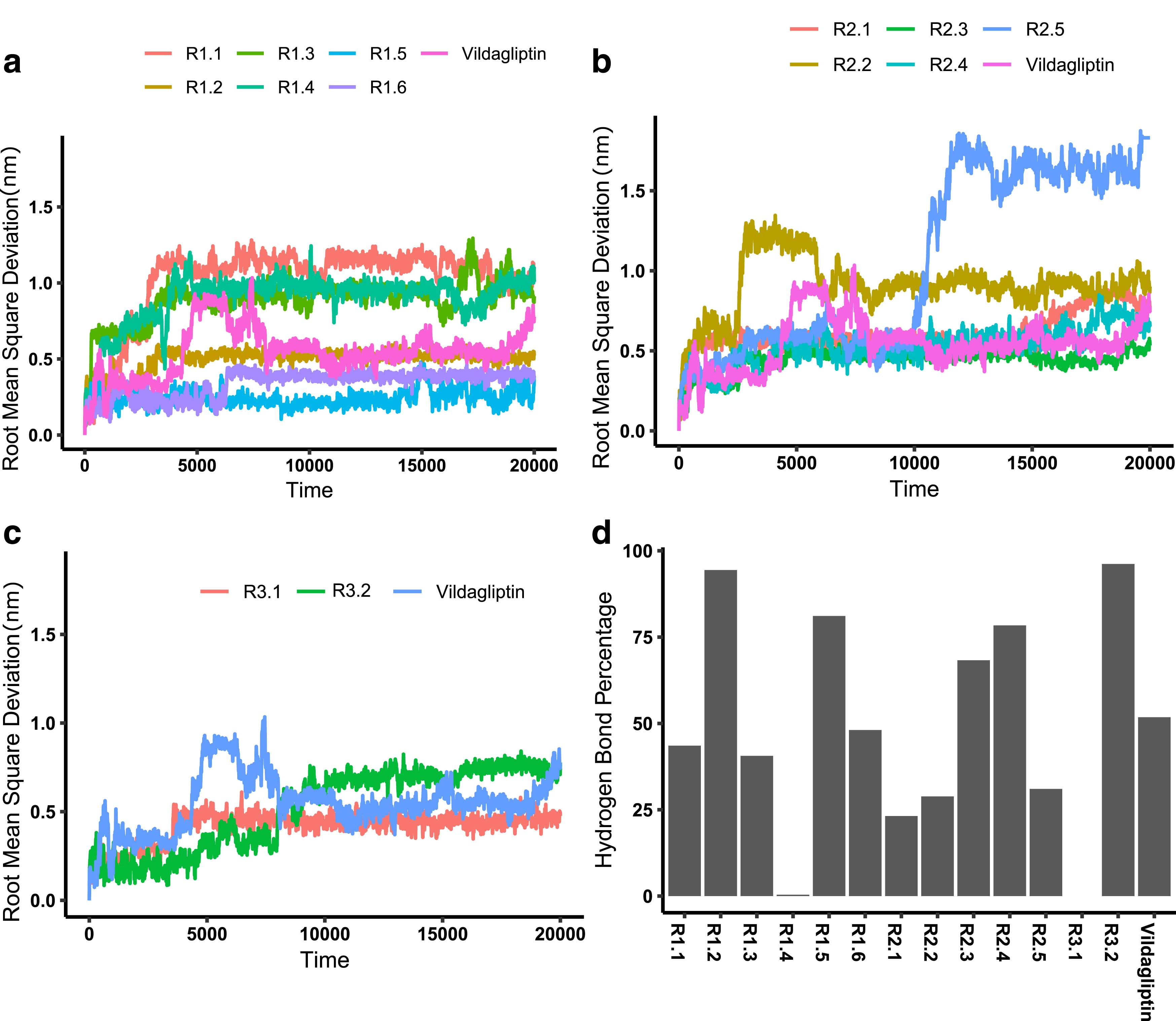
Appraisal of ligand dynamics and ligand/protein hydrogen bonding in 20 ns MD simulations of solvated DPP-4:ligand complexes for the 13 compounds identified in virtual screening as well as vildagliptin.
The formation of hydrogen bonds between the ligand and DPP-4 is an important contributor to the stability of the resultant complex. Analysis of the MD trajectories for the occurrence of at least one hydrogen bond per frame reveals that the 13 identified compounds fall into three different groups (Fig. 2d).
MM/PBSA binding energy and identification of residues involved in ligand binding
The binding energy for each of the 13 compounds identified in virtual screening was calculated using the Poisson/Boltzmann surface area continuum solvation for the full simulation time of 20 ns (Supplementary Fig. S2). The mean binding energy and its individual terms, derived from the trajectories for each compound, are summarized in Table 2.
Average MMPBSA Binding Energies (kcal mol−1) of 13 Dipeptidyl Peptidase-4-Compound Complexes Over 20 ns Simulations
Average MMPBSA Binding Energies (kcal mol−1) of 13 Dipeptidyl Peptidase-4-Compound Complexes Over 20 ns Simulations
SASA, solvent accessible surface area.
The binding energies of compounds identified by DPP-4 inhibitor-derived filters (Run 1, Run 2) range between −12 and −26 kcal mol−1 and are thus similar to that of the known inhibitor vildagliptin (−23 kcal mol−1). In contrast, the binding energies of the two compounds identified by pharmacophore search (Run 3) are significantly higher (−181 and −195 kcal mol−1), owing to an electrostatic interaction. Furthermore, evaluation of the contributions of individual residues to the binding energy identified those that most prominently engage with individual ligands. The 10 residues with largest contributions to the estimated binding energy of each ligand in the MD simulations are summarized in Table 3. This analysis shows that all 13 compounds and vildagliptin interact with glutamate-206, and that residues contributing to ligand binding are mainly located in the N-terminal recognition region (Glu204, Glu205, Glu206) and the S2 substrate binding subsite (Phe357, Tyr547) or belong to the catalytic triad (Ser630, Asp708, His740), while in
Top 10 Residues as per MMPBSA Binding Energy Contributions for 13 Compounds Complexed with Dipeptidyl Peptidase-4 from 20 ns Simulations
Known DPP-4 inhibitor.
Following screening and validation of compounds identified using the in silico pipeline discussed above, the selected compounds were resourced from commercial vendors for experimental testing. Where compounds were not available from the commercial vendor, the closest possible analogs were sought. Analogs were selected based on chemical similarity (computed using Tanimoto scoring) with a cutoff value ≥70%. No valid analog was available for
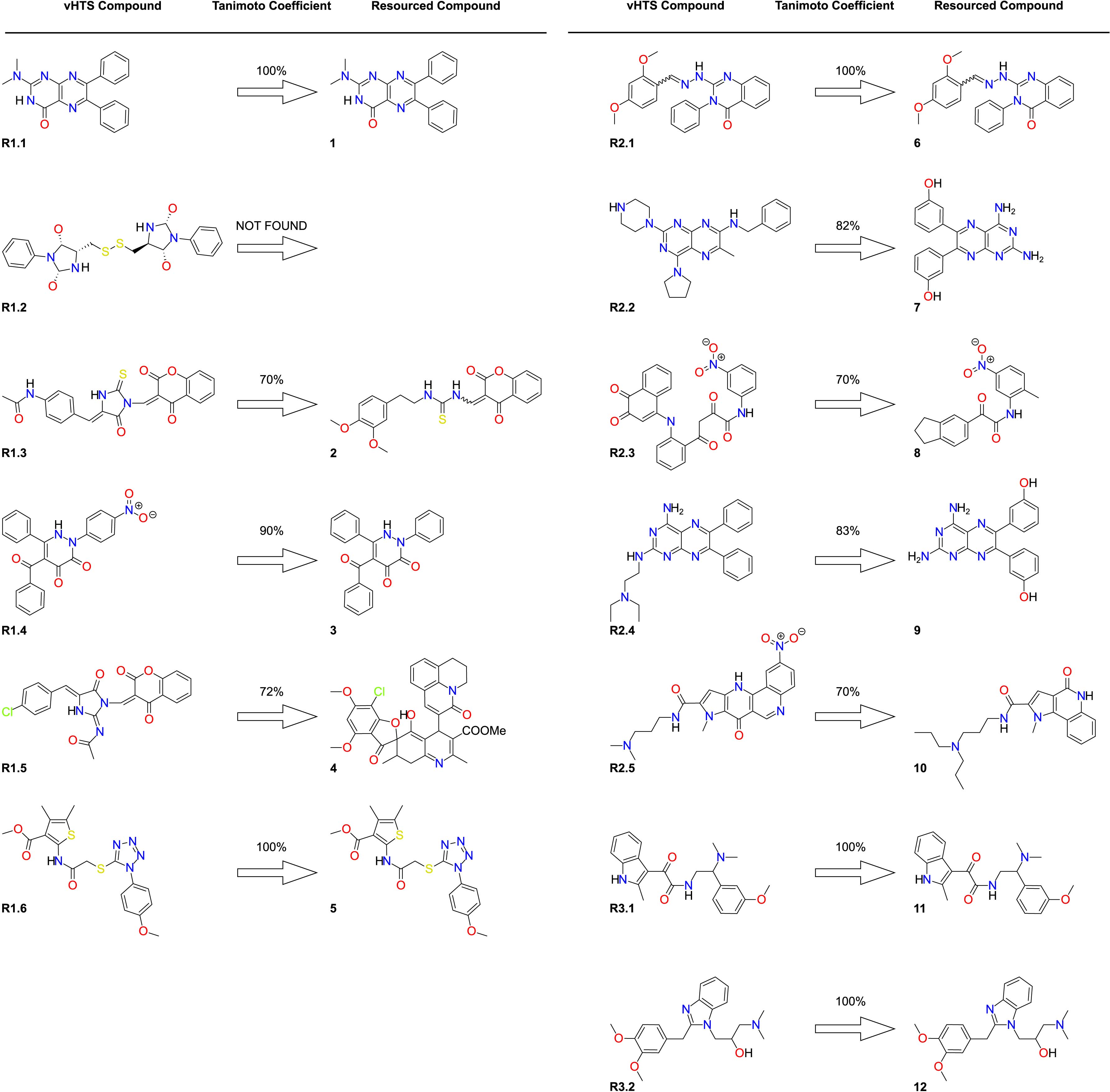
Overview of compounds screened from all vHTS runs
Of those purchased, five compounds were identical to the original vHTS hits and are shown in Figure 3 as
The purchased compounds that differed from their corresponding screening hits were subjected to 20 ns MD simulations (compounds

MD behavior of 12 purchased molecules that showed less than 100% similarity to the original hit compound under 20 ns explicit solvent conditions.
Following in silico studies, the screening hits (or analogs thereof) were tested for binding to DPP-4 using an in vitro enzyme inhibition assay from the DPP-IV Drug Discovery Kit (Enzo Life Sciences). In this study, the inhibition of DPP-4 by five original hit compounds (
As shown in Figure 5a, 2 and 4 had the greatest inhibitory effects and lowered the activity of DPP-4 by 23% and 22%, respectively; the corresponding decreases in DPP-4 activity in the presence of

In vitro testing of screened compound at
Based on the structure of fragment 1 of most active molecule

Spirochromanone compounds.
The temperature and pressure of NVT and NPT ensembles in the 100 ps equilibrium phase and the total potential energy curves of the 20 ns simulation phase are shown in Supplementary Figure 3. These five compounds were tested in vitro using against DPP-4 at 100 μM and one compound (
Overall, this study identified two compounds capable of reducing the activity of DPP-4 by 50% at 100 μM. Both possess novel spirochromanone centers not previously seen in DPP-4 inhibitors, although they show similar binding patterns to anagliptin and sitagliptin. These molecules have potential as lead compounds for the development of analogs with greater inhibitory activity against DPP-4.
This study demonstrates the efficacy of computer-aided drug design for the discovery of novel inhibitors of DPP-4, a prime target for treating type 2 diabetes and cardiovascular diseases. The present DPP-4 inhibitors were used as reference molecules to guide the structure-based screening approach. A comprehensive library of 4 million compounds was screened in silico using physicochemical filters based on the proposed binding region of DPP-4 to capture potential ligands. Similarly, the ZINC database was searched using a pharmacophore based on the cocrystal structures of DPP-4 with known inhibitors; the resultant 2000 compounds were used as a library for further virtual screening to identify those with the greatest structural complementary to the proposed binding region of DPP-4. The hit compounds were then docked in the binding site and subjected to MD simulations; all assumed stable conformations within the binding site of DPP-4 within 20 ns. MMPBSA calculations were used to predict the stability and binding affinity of hit molecules. This complete pipeline identified 13 compounds with potential binding affinity for DPP-4. As none of these compounds is more than 40% similar in structure (per Tanimoto coefficient) to the others or any known DPP-4 inhibitor, a novel scaffold for DPP-4 inhibition has been identified. The ligand having the highest scoring docked pose of all 13 compounds was found to have negative binding energy using both the AutoDock and INVENTUS docking packages.
Although they use two different algorithms, both packages produced binding scores that are well-correlated (Pearson coefficient 0.84), which supports the reliability of the predicted binding modes of these molecules. The average MMPBSA binding energies of the docked complexes for 20 ns simulations fall within the acceptable range of −26.41 to −11.88 kcal mol−1. The complexes were also assessed for the role of the protein, and the main residues involved in protein/ligand interactions were also detected. Many of these residues are already known to contribute to interactions between DPP-4 and existing inhibitors. The hit compounds (or the closest structural analogs available) were purchased and tested for DPP-4 inhibition in enzyme assays. One molecule from the set of compounds reduced DPP-4 activity to 53% at 100 μM. With a predicted binding pose that is similar to those of know inhibitors (anagliptin and sitagliptin) but a novel scaffold, this compound comprises two structural elements/fragments, one of which contains spirochromanone-type center. To investigate this scaffold further, five additional compounds with similar topology were synthesized and tested both in silico and in enzyme inhibition assays. In this study, a second compound was found to reduce DPP-4 activity to 53%.
In summary, this study has used a rapid in silico screening technique to identify two active compounds, containing a novel spirochromanone scaffold, that are able to reduce DPP-4 activity by 50%. As they are structurally distinct from existing DPP-4 inhibitors/binders, both have the potential to serve as leads for medicinal chemistry approaches to enhance and refine their activity to achieve greater inhibition and, potentially, develop novel treatments for DPP-4-associated disorders.
Footnotes
Acknowledgments
A.M. acknowledges the Department of Biotechnology (DBT), India, for the award of an Indo-Australian Gold fellowship (BT/IACBGF/03/03/2015) and support from the Institute for Integrated and Intelligent Systems, Griffith University. M.C. is supported by PhD scholarships from The Equity Trustees and the Australian Government Research Training Program.
Author Disclosure Statement
The authors declare there are no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.