
Abstract
This investigation aimed to explore the underlying prognosis-associated microRNA (miRNA) biomarkers in endometrial cancer. Homo sapiens miRNA data set GSE35794 and miRNA data in TGGA database were downloaded and applied to screen the differentially expressed miRNAs (DE-miRNAs) using unpaired t-test in limma package in R. Basing on Venn analysis, the overlapped DE-miRNAs were screened and their potential targets were predicted according to miRWalk followed by target functional enrichment analyses and protein–protein interaction network visualized using Cytoscape. Finally, according to the information provided by the The Cancer Genome Atlas (TCGA) database, correlations between miRNAs or targets and patient prognosis were analyzed by survival package in R. A total of 24 overlapped DE-miRNAs were identified between endometrioid endometrial cancer samples and normal samples. Then, the miRNA-target regulatory network was constructed, including 11 upregulated miRNAs (e.g., miR-200a, miR-200b, and miR-200c) and five downregulated miRNAs (e.g., miR-449a, miR-145-5p, and miR-145-3p). Lymphocyte enhancer factor-1 (LEF1) was predicted to be a target of miR-449a and SOX11 was a target of miR-145-5p. Functional enrichment analyses of these targets were significantly related to the biological process of “negative regulation of transcription from RNA polymerase II promoter” and “positive regulation of transcription from RNA polymerase II promoter” (e.g., NOTCH1, LEF1, and SOX11). In addition, survival analysis showed that miR-449a, miR-145-5p, and LEF1 were approximately correlated with the overall survival prognosis of endometrial cancer patients. Downregulations of miR-449a and miR-145-5p might be involved in the pathogenesis of endometrial cancer and could act as prognostic biomarkers for endometrial cancer patients.
1. Introduction
Endometrial cancer is one of the common gynecologic cancers in developed countries and the second most common in developing countries, second by cervical cancer (Plaxe and Mundt, 2015). Although the incidences of endometrial cancer in Asian nations, such as China, Japan, and Korean, are lower than that in the Western developed countries, the incidence of endometrial cancer in China has increased significantly in recent years (Sun et al., 2018). In some developed areas of China, endometrial cancer has supplanted cervical cancer to be the top one among gynecologic cancers (He et al., 2018). Although great efforts have been made, prognosis of endometrial cancer patients remains to be improved (Svanvik et al., 2017). Therefore, it is of importance to further explore the mechanism of endometrial cancer to improve the diagnosis and treatment of endometrial cancer.
MicroRNAs (miRNAs), a class of single-stranded noncoding RNA molecules with length of 19–25 nt, are considered to be responsible for the progression of endometrial cancer (Torres et al., 2013b), including cell cycle progress, proliferation, migration, and invasion in endometrioid endometrial cancer (EEC; Chung et al., 2012; Qin et al., 2012; Xu et al., 2013). For example, miR-15a-5p inhibits the growth of endometrial cancer cells via attenuating WNT3A expression in the Wnt/β-catenin signaling pathway (Wang et al., 2017). Downregulation of miR-125b promotes the invasion of endometrial cancer via targeting ERBB2 (Shang et al., 2012). Overexpression of miR-204 could repress the migration and invasion of endometrial cancer cells via targeting FOXC1 (Chung et al., 2012). Dong et al. (2014) have summarized that miRNAs could also regulate the progress of epithelial/mesenchymal transition via targeting Twists1, ZEB1, and BMI-1 in the PI3K/AKT signaling pathway to modulate the pathogenesis of endometrial cancer. In addition, miRNAs, such as miR-135b and miR-205, could be used for early detection and follow-up estimation of endometrial cancer (Tsukamoto et al., 2014). However, these findings are still not enough to clarify the mechanism of endometrial cancer.
To get an overall understanding of miRNAs in EEC, Torres et al. (2013a) have exploited the GSE35794 data set to screen several differentially expressed miRNAs (DE-miRNAs) between EEC tumor samples and normal samples used for the diagnostic and prognostic biomarker of endometrial cancer. However, the detailed mechanisms of these DE-miRNAs involved in endometrial cancer remain unexplored. In this study, the regulatory mechanisms between DE-miRNAs and their targeted genes were analyzed to explore the underlying molecular mechanisms in endometrial cancer to provide some new insights in the treatment of endometrial cancer.
2. Methods
2.1. Data source
Homo sapiens miRNA data set GSE35794 was downloaded from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35794), which includes 17 endometrial cancer samples and four healthy control samples. All samples included in this data set were sequenced on the platform of GPL10850 Agilent-021827 Human miRNA Microarray (V3; miRBase release 12.0 miRNA ID version). In addition, the matrix miRNA data set of endometrial cancer, as well as the clinical information on The Cancer Genome Atlas (TCGA) uterine corpus endometrial carcinoma (UCEC), was also downloaded from the TCGA database, including 177 tumor tissue samples and 24 adjacent normal controls.
2.2. Data preprocessing
For raw data downloaded from GEO data sets, limma package (version 3.10.3, www.bioconductor.org/packages/2.9/bioc/html/limma.html; Smyth, 2011) in R (version 3.3.2) was used for normalization and background correction. For the data set in TCGA database, all matrix files have been preprocessed.
2.3. DE-miRNA screening
According to sample information, gene expression matrix was divided into the tumor and control groups and DEGs between groups were identified by unpaired t-test in limma package with the thresholds of p < 0.05 and |log fold change (FC)| >2. Following this, overlapped DE-miRNAs between the GEO data set and TCGA data set were screened via Venn analysis and used for the following investigation.
2.4. Construction of miRNA-target gene regulatory network
Based on miRWalk2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2; Dweep and Gretz, 2015), miRNA-target gene regulatory pairs predicted by miRWalk, miRanda, miRDB, miRMap, miRNAMap, Pictar2, RNA22, and TargetScan were selected to construct the miRNA-target gene regulatory network by Cytoscape (version 3.2.0, www.cytoscape.org; Shannon et al., 2003).
2.5. Functional enrichment analyses of miRNA-targeted genes
Based on miRNA-target regulatory network, genes involved in this network were subjected to the Gene Ontology biology process (GO-BP; Ashburner et al., 2000) and Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa and Goto, 2000) enrichment analyses with the Database for Annotation, Visualization, and Integrated Discovery online tool (version 6.8, https://david.ncifcrf.gov; Huang et al., 2008a,b). Significant enrichment results were considered when p < 0.05 and gene count >2.
2.6. Construction of protein–protein interaction network
Following miRNA-targeted genes had been predicted, interactions among gene-encoded proteins had also been predicted using STRING database (version 10.0, www.string-db.org; Szklarczyk et al., 2014) with median confidence = 0.4. Subsequently, the interactions between proteins were visualized using Cytoscape (version 3.2.0). Degree centrality was used to assess the interaction relationship. The higher the score, the more important the node (protein) presented in the network.
2.7. Survival analysis
According to the expression, miRNAs and proteins were divided into high and low expression according to their median expression. Then, survival package (version 2.42-6, http://cran.r-project.org/web/packages/survival/index.html; Therneau and Lumley, 2009) in R software was used to analyze the association between miRNA expression and patient's overall survival (OS) according to the information provided by the TCGA UCEC.
3. Results
3.1. Screening the DE-miRNAs between endometrial cancer samples and healthy controls
After analyzing, a total of 37 DE-miRNAs were identified between endometrial cancer samples and healthy controls in GSE35794, including 16 upregulated miRNAs and 21 downregulated miRNAs. Moreover, in TCGA-UCEC miRNA matrix data, a total of 121 DE-miRNAs were identified with the same criteria, including 102 upregulated miRNAs and 19 downregulated miRNAs. Then, the overlapped miRNAs between these two data sets were analyzed using VENN analysis. As a result, a total of 24 overlapped DE-miRNAs were screened for the following investigations, including 15 upregulated DE-miRNAs and 9 downregulated DE-miRNAs (Fig. 1).

Overlapped miRNAs screened by Venn analysis between TCGA database and GSE35794 data set.
3.2. miRNA-target gene regulatory network
According to miRWalk2.0, a total of 205 miRNA-target pairs were identified and subjected to construct an miRNA-target regulatory network (Fig. 2). In this network, a total of 10 upregulated miRNAs were involved, such as miR-141-3p, miR-200a-3p, miR-200c-3p, and miR-183-5p, and six downregulated miRNAs, including miR-449, miR-381-3p, miR-143-3p, miR-542-3p, miR-145-5p, and miR-133b. Specifically, miR-449a was a downregulated DE-miRNA and predicted to the maximum targets, including LEF1, NOTCH1, and NRIP3. miR-145-5p was another downregulated DE-miRNA and predicted to target MYO5A, SOX11, and RTKN.
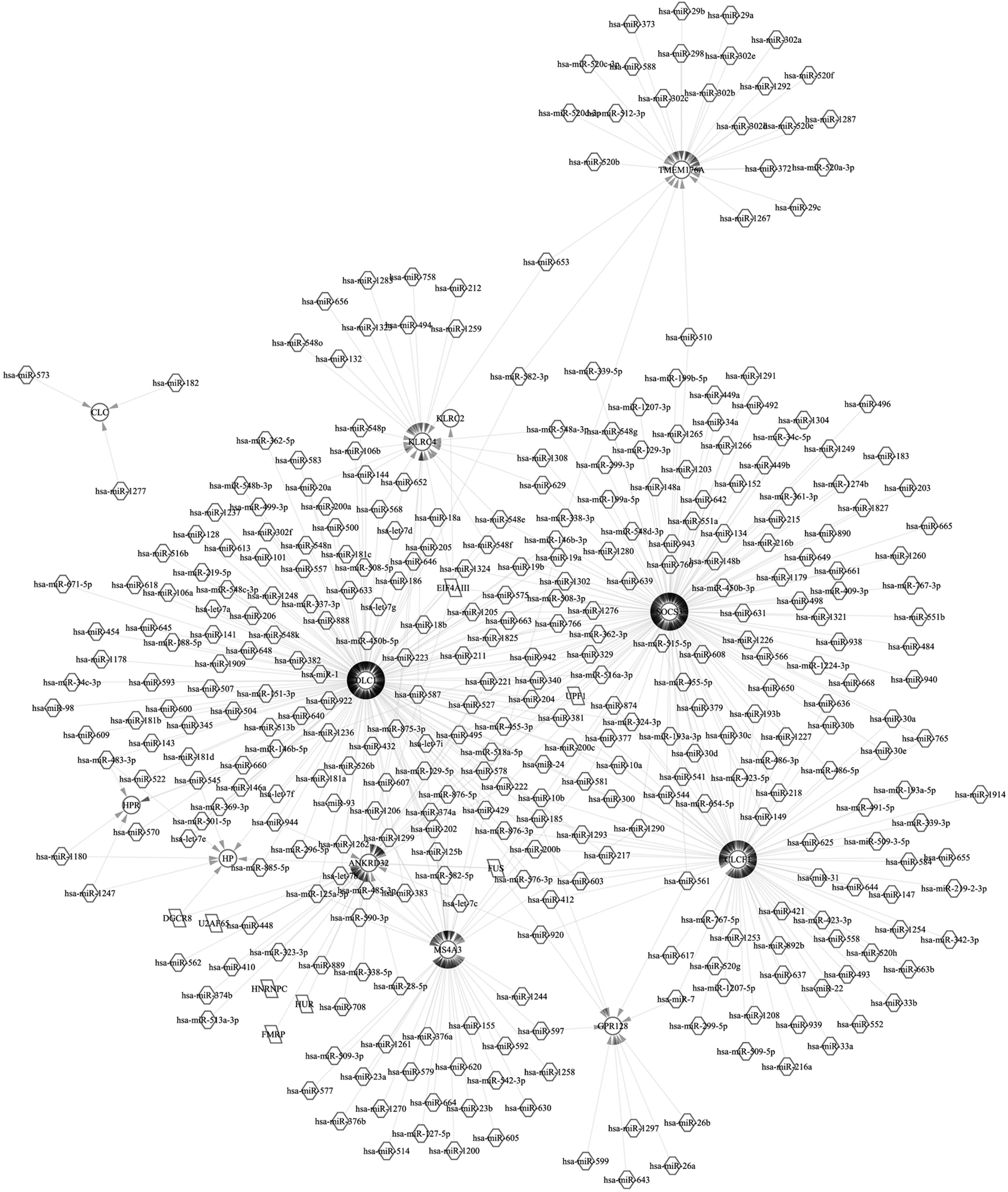
miRNA-target regulatory network. Triangle represents the upregulated miRNA, arrow represents downregulated miRNA, and circle represents the targeted gene.
3.3. Functional enrichment analysis for miRNA-targeted genes
Following the investigation, miRNA-targeted genes were subjected to GO-BP and KEGG functional enrichment analysis. As a result, the upregulated miRNA-targeted genes were significantly enriched in five KEGG pathways, including the oxytocin signaling pathway (p = 0.016), phosphatidylinositol signaling system (p = 0.025), miRNAs in cancer (p = 0.037), proteoglycans in cancer (p = 0.041), and Rap1 signaling pathway (p = 0.047); however, only one KEGG pathway was significantly enriched by downregulated miRNA-targeted genes, namely arrhythmogenic right ventricular cardiomyopathy (p = 0.016; Fig. 3). Moreover, a total of 42 GO-BPs were enriched by the upregulated miRNA-targeted genes, such as negative regulation of transcription from RNA polymerase II promoter (p = 1.54 × 10−4), positive regulation of transcription from RNA polymerase II promoter (p = 3.77 × 10−4), positive regulation of cell proliferation (p = 2.58 × 10−3), protein ubiquitination involved in the ubiquitin-dependent protein catabolic process (p = 2.73 × 10−3), and positive regulation of cholesterol efflux (p = 0.0034). In addition, a total of 19 GP-BPs were enriched by the downregulated miRNA-targeted genes, including positive regulation of transcription from RNA polymerase II promoter (p = 2.82 × 10−5), DNA templated (p = 8.81 × 10−4), chemical synaptic transmission (p = 0.006), histone H3 acetylation (p = 0.0075), and protein localization to plasma membrane (p = 0.016; Fig. 3).
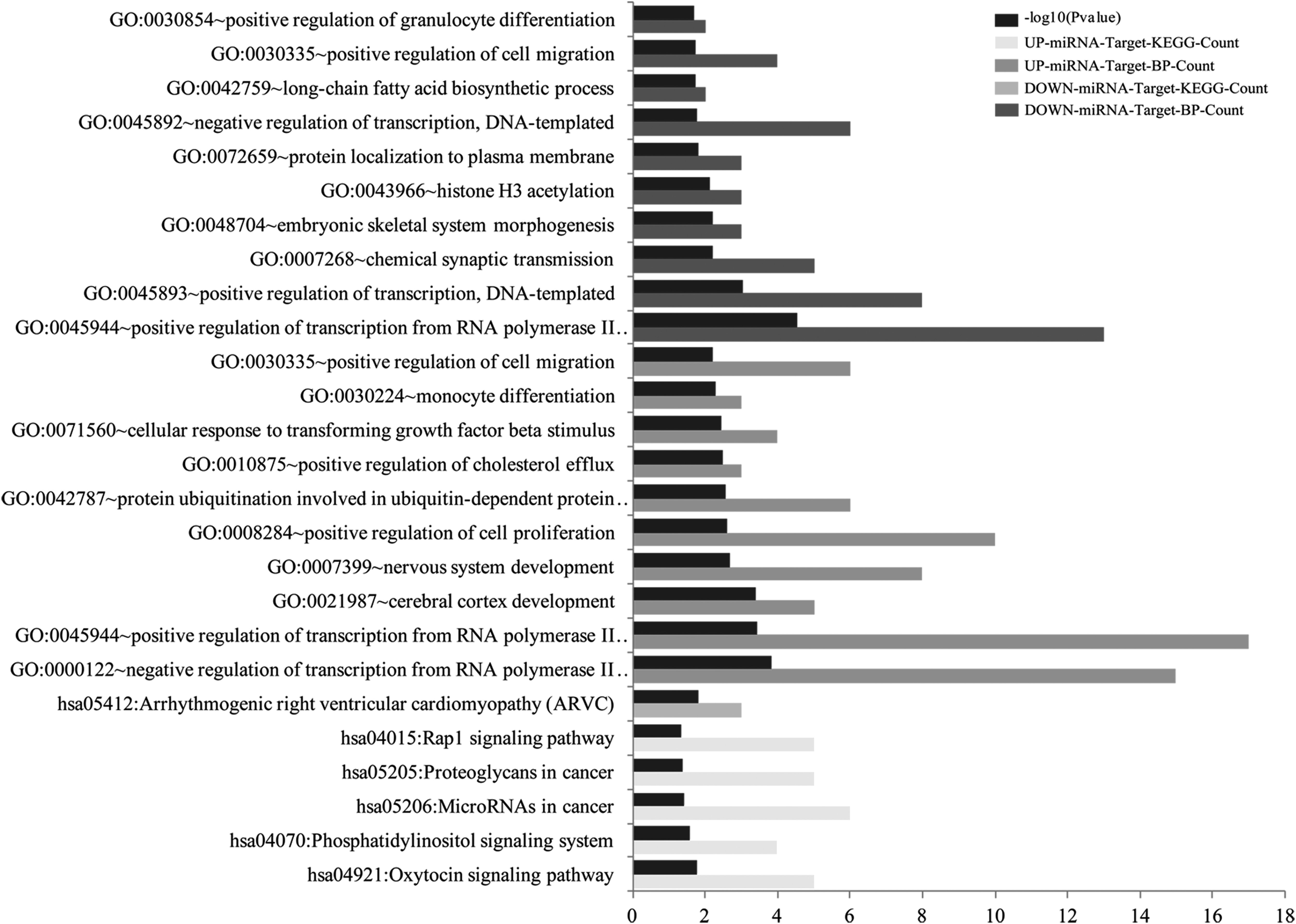
Functional enrichment analysis for miRNA-targeted genes. BP, biology process; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
3.4. Protein–protein interaction network
Furthermore, the interactions between miRNA-targeted gene-encoded proteins were predicted by STRING database and visualized using Cytoscape (Fig. 4). A total of 106 nodes (proteins) and 159 interaction pairs were included. Specifically, the top 20 nodes with the high interaction pairs in turn were: PIKFYVE, VEGFA, KAT2B, NOTCH1, KDR, SIRT1, MEF2C, SYNJ1, UBE2B, DUSP1, MYO1C, DUSP1, UBE2E1, ITPR1, DDX17, SH3KBP1, LEF1m RALA, PLCB1, and KAT6B.

Protein–protein interaction network for miRNA-targeted gene-encoded proteins. Circle represents the target of upregulated miRNA, and diamond represents the targets of downregulated miRNA. The bigger the node size presented, the higher the degree the node has.
3.5. Survival analysis
According to the expression median value, miRNA and top 20 nodes in protein–protein interaction network were divided into high and low expression. Then, the association between gene expression and patients’ OS prognosis was assessed and found that LEF1 was significantly correlated with the OS of patients and miR-449a and miR-145-5p might approximately correlate with the OS of patients (Fig. 5).

Overall survival analysis for miRNA and its targeted genes.
4. Discussion
In the present study, a total of 103 upregulated and 18 downregulated DE-miRNAs were identified between endometrial cancer samples and control samples. Among these DE-miRNAs, a total of 11 upregulated DE-miRNAs and five downregulated DE-miRNAs were predicted to construct the miRNA-target regulatory network, including the regulatory relationships of miR-449a-LEF1, miR-449a-NOTCH1, and miR-145-5p-SOX11. Functional enrichment analysis of these miRNA-targeted genes revealed that DE-miRNA-targeted genes were significantly related to the biological process of “negative regulation of transcription from RNA polymerase II promoter” and “negative regulation of transcription from RNA polymerase II promoter” (e.g., NOTCH1, LEF1, and SOX11). In addition, survival analysis showed that miR-449a, miR-145-5p, and LEF1 were approximately correlated with the OS prognosis of endometrial cancer patients.
miR-449a is previously reported as a tumor suppressor in several cancers. For example, miR-449a could promote apoptosis of liver cancer cell via downregulating the expression of calpain 6 and POU2F1 (Liu et al., 2016), and it could also inhibit the epithelial/mesenchymal transition and metastasis via targeting several downstream genes during hepatoma carcinogenesis (Chen et al., 2015). miR-449a could also serve as a tumor suppressor in gastric cancer via inhibiting the expression of E2F3 to suppress proliferation and induce apoptosis of cancer cells (Li et al., 2015). In addition, miR-449a inhibits the pathogenesis of glioblastoma via targeting Myc-associated zinc-finger protein (Yilong et al., 2015). These findings indicated that miR-449a might play critical role in endometrial cancer. In the present study, miR-449a was identified to be significantly downregulated in endometrial cancer, and high expression of miR-449a is obviously correlated with better prognosis of patients with endometrial cancer within 120 months, suggesting that miR-449a could act as a prognostic biomarker for endometrial cancer. Further functional enrichment analysis revealed that miR-449a-targeted genes were significantly enriched in the biological process of transcription from RNA polymerase II promoter, suggested that miR-449a might regulate the endometrial cancer progression via modulating transcription-associated gene expression.
LEF1, the code of lymphocyte enhancer factor-1 (LEF-1), is a major transcription factor of Wnt pathway and has been reported to play a critical role in cancer development (Xingchun et al., 2014). miR-34a could target downregulating the expression of LEF1 to inhibit the proliferation and invasion of prostate cancer cells (Liang et al., 2015). Shelton et al. (2012) have reported that LEF1 protein expression was significantly higher in endometrial cancers within mice and human compared with the normal endometrium. However, LEF1 was identified to be a target of miR-449a, which was significantly downregulated in endometrial cancer in this study. In a previous study, Seungil et al. (2012) have demonstrated that miR-449a could modulate chondrogenesis of human mesenchymal stem cells via directly targeting LEF1. Further survival analysis showed that patients with higher expression of LEF1 present better prognosis than those with lower expression. Previous study showed that LEF1 could function as transcriptional inhibitors of estrogen receptor activity in breast cancer, suggesting that LEF1 was involved in hormone response-associated cancers (Holmes et al., 2008). Thus, we suspected whether there were some correlations between estrogen levels and LEF1 function in endometrial cancer, to modulate the development of endometrial cancer. Further mechanisms of investigations about miR-449a-targeted LEF1 are required in endometrial cancer.
miR-145-4p is a common noncoding RNA involved in the pathogenesis of several cancers. For example, Eades et al. (2015) have identified that miR-145 modulates the invasion of cancer cells via targeting ARF-6 in triple-negative breast cancer. Overexpression of miR-483-3p overcomes the effect of miR-145/TP53 proapoptotic loop in hepatocellular carcinoma (Lupini et al., 2016). Besides, Wang et al. (2015) have revealed that miR-145, miR-125a, and miR-146a could act as diagnostic biomarkers for nonsmall-cell lung cancer. In the present study, miR-145-5p was significantly downregulated in endometrial cancer and patients with lower expression of miR-145-5p presented poor prognosis than those with a higher expression of miR-145-5p, suggesting that miR-145-5p might act as a tumor suppressor in the development of endometrial cancer. Yanjing et al. (2011) have revealed that upregulating the expression of miR-145 could promote the differentiation of endometrial adenocarcinoma cells via targeting OCT4. In the current study, we had also identified that miR-145-5p could target SOX11 which was significantly enriched in positive regulation of transcription from RNA polymerase II promoter and positive regulation of transcription, DNA templated biological processes. Therefore, we deduced that miR-145-5p might regulate cell cycle-associated processes to inhibit the development of endometrial cancer and could serve as a prognostic biomarker for patients with endometrial cancer.
There are still some limitations in this study. First, multiple abnormal miRNAs have been identified to be associated with endometrial cancer in the present study, but the associated information was obtained in silico and the detailed mechanisms involved in the pathogenesis of endometrial cancer still need further exploration. Second, miR-449a is an onco-miRNA in endometrial cancer, controversial to other cancers and required further validation. Last, although LEF1 was identified to be a target of miR-449a and positively correlated with the prognosis of patients with endometrial cancer, further clinical confirmation is still required.
In conclusion, miR-449a and miR-145-5p were significantly upregulated in endometrial cancer and high expression levels of miR-449 and miR-145 were obviously correlated with better prognosis of patients with endometrial cancer. LEF1 was predicted to be a target of miR-449a and significantly correlated with the prognosis of patients with endometrial cancer. However, the mechanisms of investigations about the miR-449a-targeted LEF1 are required in endometrial cancer in the future.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.